iGEM REPORT: Leijuvant-A Revolutionary Choice of Vaccine Helper; Construction of an E. coli-Leishmania shuttle vector
Note: This iGEM Report was submitted to the PLOS iGEM Realtime Peer Review Jamboree, and has not undergone formal peer review by any of the PLOS journals. We welcome your comments on this work.
Leijuvant-A Revolutionary Choice of Vaccine Helper; Construction of an E. coli-Leishmania shuttle vector
Chia-Wei Chang (1), Chien-Yu Hsu (1), Justine Hsu (1), Yu-Chang Ku (1), Tzu-Chieh Liao (1), Bin-Tse Lin (1), Tzu-Li Liu (2), Min Lu (1), Ying-Hsuan Lu (1), Yu-Yan Wang (1)*, Da-Li Yen (1), Kwang-Poo Chang (3), Guang-Wu Chen (4), Chi-Ching Lee (4), Chao-Lan Yu (1)
- Department of Biomedical Sciences, College of Medicine, Chang Gung University, Taoyuan, Taiwan.
- Department of Electrical Engineering, College of Engineering, Chang Gung University, Taoyuan, Taiwan.
- Department of Microbiology and Immunology, Chicago Medical School, Rosalind Franklin University of Medicine and Science, North Chicago, Illinois, United States of America.
- Department of Computer Science and Information Engineering, College of Engineering, Chang Gung University, Taoyuan, Taiwan.
*Corresponding author: Yu-Yan Wang ([email protected])
Author Contributions
Conceptualization: CWC, CYH, JH, YCK, TCL, BTL, TLL, ML, YHL, YYW, DLY
Investigation: CWC, JH, YCK, YYW, DLY
Resources: KPC
Writing – Original Draft: CWC, CYH, JH, YCK, TCL, BTL, ML, YHL, YYW, DLY
Writing – Review & Editing: YYW
Visualization: CWC, YCK, YYW
Funding Acquisition: BTL
Supervision: KPC, GWC, CCL, CLY
Abstract
For many infectious diseases that still don’t have an effective vaccine, enhancing T cell immune response may be the key to solve this problem. Leijuvant represents a whole new perspective of adjuvant that uses Leishmania as an effective T cell stimulator. Leishmania is a parasite that specifically lives within macrophage, a professional antigen presenting cell (APC). As a potential vaccine adjuvant, Leishmania possesses many advantages, including APC recruitment, activation of innate immunity, activation of antigen presentation, and most important of all, T cell activation. Genetically-engineered Leishmania that can be inactivated by light exposure acts as a safe carrier to deliver specific antigens to APCs for activation of T and B cells. Based on this concept, we established a new model system to generate antigen-specific Leishmania adjuvant – Leijuvant. Our ultimate goal is to introduce Leijuvant as an effective, safe, and antigen-specific adjuvant to the vaccine industry and the general public. For the beginning, we built a shuttle vector that can express proteins in Escherichia coli and Leishmania. We used this shuttle vector to express hemagglutinin of H1N1 influenza virus and ovalbumin in Leishmania. After drug selection, we analyzed protein expression of stable Leishmania transfectants by immunoblotting. The absence of hemagglutinin and ovalbumin expression suggests the importance of a 2.3-kb Leishmania intergenic sequence in building an effective shuttle vector.
Financial Disclosure
Funding was provided by GeneDireX, Body Organ Biomedical Corporation (BOBC), Integrated Device Technology (IDT), Taqkey Science, and Chang Gung University. However, the funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
The authors have declared that no competing interests exist.
Data Availability
All data are fully available without restriction: http://2016.igem.org/Team:CGU_Taiwan.
Introduction
Vaccines save millions of lives each year and are among the greatest achievements of biomedical science and the most cost-effective health interventions ever developed [1-3]. Endemic transmission of poliovirus, measles, and rubella viruses has been eliminated in the United States. Smallpox has been eradicated worldwide[4]. By improving vaccines, many more lives can be saved each year.
There are still a number of severe diseases that don’t have an effective vaccine, such as hepatitis C, tuberculosis, and malaria [5,6]. Researchers concluded that the reason why most of the vaccines against these diseases failed to stimulate effective immunity is due to the lack of T cell immune response [7]. For many infectious diseases, T cells play an important role in naturally acquired protective immune response [8]. T cells are activated by antigen presenting cells (APCs), such as macrophage or dendrite cell. Activated T cells then trigger both humoral and cell-mediated immune responses. T cells are essential to the induction of high-affinity antibodies and immune memory [9]. Optimizing T cell response by vaccination has been the aim of many scientists and is thought to be the future of vaccine development [10,11].
Many vaccines require immunologic adjuvant to stimulate an effective immune response [12]. Adjuvants are compounds that can improve immunogenicity of vaccines by triggering the innate immune response to accelerate, prolong, or enhance antigen-specific immune response [13]. Despite the success of current adjuvants Alum and MF59, there is still a need for further improvement [14-17]. Specifically, the challenge of vaccine industry is to make adjuvant stimulating stronger T cell response and antibody production for protective immunity.
Leishmania is a genus of trypanosoma protozoa that are responsible for leishmaniasis, a blood-borne disease spread by blood-sucking sandflies [18]. Leishmania multiply and develop extracellularly as promastigotes in the digestive tract of the sandflies. When the female fly takes blood meals, the promastigotes are delivered into the skin of the mammalian host. They will directly infect macrophages where they differentiate into amastigotes and multiply intracellularly [19].
Leishmania are aerobic organisms that rely on oxidative phosphorylation, but are defective in the synthesis of heme which is required for electron transport respiratory complexes. The genetic deficiency of heme biosynthesis in Leishmania makes it possible to produce transgenic DT mutants, which are inducible with delta-aminolevulinate (ALA) for accumulation of uroporphyrin I (URO) as a photosensitizer. Photosensitizers like URO and aluminum phthalocyanine (PC) can be excited when illuminated at specific wavelengths and produce singlet oxygen and reactive oxygen species (ROS) to kill these parasitic protozoa (oxidative inactivation) [20,21]. Professor Kwang-Poo Chang established this double-photo inactivation system of Leishmania. He adapted a combinational approach by loading the Leishmania DT mutants both endogenously with URO via the use of ALA and exogenously with PC. After URO and PC are illuminated by specific wavelengths of light, double inactivation kills Leishmania with proven effectiveness. He also validated the role of photo-inactivated Leishmania in modulating immune response [22].
This photodynamic Leishmania system has two major advantages to function as an immunologic adjuvant. First, it can be specifically recognized by APCs for delivery of target antigens with higher efficiency. So, it may stimulate stronger immune response and enhance antibody production. Second, photo-inactivation of Leishmania provides a safe platform for vaccine development. Photo-inactivation of Leishmania can be verified by loss of flagellar motility under microscopy and no growth in culture to ensure safety as adjuvant [23]. These two advantages suggest that photo-inactivated Leishmania can be a novel system of immunologic adjuvant for next-generation vaccination. Therefore, we plan to establish a new model system in the context of biobricks to generate antigen-specific Leishmania adjuvant as a T cell stimulator – Leijuvant.
Materials and Methods
Basic molecular biology techniques
All detailed protocols used in this work, including basic molecular biology techniques and cloning, are available online (http://2016.igem.org/Team:CGU_Taiwan/Notebook).
Detailed information of BioBrick constructions are also available through http://2016.igem.org/Team:CGU_Taiwan/Parts.
Cell cultures
We used wild-type clone 12-1 of Leishmania amazonensis (RAT/BA/74/LV78) which was doubly transfected with pX-alad and p6.5-pbgd (DT; 8–10). ALAD and PBGD are second and third enzymes in the heme biosynthetic pathway, respectively. Leishmania were routinely grown as promastigotes at 20°C in medium 199 (Sigma) supplemented with 10% heat-inactivated fetal bovine serum (HIFBS) and 25 mM HEPES (pH 7.4). G418 (100 μg/ml) and tunicamycin (20 μg/ml) were added to maintain ALAD and PBGD expression. Cells were passaged in a week when the growth reached the peak cell density of about 5×107 cells/ml at the stationary phase. Before exposure of these DT cultures to ALA for inducing cytoplasmic accumulation of URO, they were grown in drug-free medium to stationary phase to avoid potential cytotoxicity of the drugs carried over to the host cells.
Transfection of Leishmania promastigotes by electroporation
Leishmania were grown to late-log phase (5-10×107 cells /ml) in 4 ml culture. Cells were pelleted at 3,500x g at 4°C for 5 min and washed twice with ice cold transfection buffer. Washed cells were resuspend in transfection buffer to a final density of 108 cells/ml and kept on ice. About 15-20 µg of plasmid DNA (in 20-30 µl) were added into 300 µl of cell suspension and chilled on ice for 10 min. About 320-330 µl of DNA-cell mixture were transferred into pre-chilled 0.2 cm Biorad E cuvette. Electroporation was set at 0.45 kV and 500 µF for 4-6 msec. Electroporated cells were immediately transferred to 3 ml of medium199 with 20% HIFBS for recovering at 25°C for 3-24 hr.
Drug selection of Leishmania transfectants
Different concentrations of antibiotic were added into recovered cells to start selection of stably transfected cells. Initial hygromycin concentrations started at 5 and 10 µg/ml. After cell growth to full turbidity, cells were exposed to increasing concentrations of hygromycin from 50, 100, 250, to 500 µg/ml.
Western blot analysis
Leishmania transfectants were pelleted by centrifugation at 3,500x g at 4°C for 5 min. Cells were lysed in 1X SDS sample buffer at a concentration of 10 µl SDS sample buffer per 107 cells. As a positive control for hemagglutinin (HA), whole cell lysates were prepared from HA-expressing 293T cells in NP40 lysis buffer. Albumin from chicken egg white (Sigma-Aldrich, A5503) was diluted to 10 ng/µl and 10 ng was loaded as a positive control for ovalbumin (OVA). Proteins were resolved by SDS-PAGE, transferred to nitrocellulose membrane, and then detected by immunoblotting using specific antibodies. Primary antibodies include anti-OVA (Abcam 6C8, 1:1000), anti-HA (GeneTex GTX127357, 1:3000), and anti-tubulin (Millipore, 05-661, 1:10000). Antibody specific for Leishmania p36 protein was a generous gift from Professor Kwang-Poo Chang. Appropriate secondary antibodies conjugated with horseradish peroxidase were used to detect signals by enhanced chemiluminescence system.
IPTG induction of HA protein
For IPTG-induced expression of HA in bacteria, we built pSB1C3-J04500-HA and transformed it into E. coli BL21. A second construct pSB1C3-J04500-MAL-p5x-HA was made by ligating pMAL-p5x-HA and pSB1C3-J04500. Detailed information about pMAL-p5x-HA can be found on the following website: https://biomod2016.gitlab.io/cgu/. To induce HA protein, IPTG was added into 10 ml of transformed BL21 to a final concentration of 0.2 mM. For uninduced control, ZnSO4 was added into 10 ml of transformed BL21 to a final concentration of 10 μM. Cells were lysed with 1X SDS sample buffer and analyzed by immunoblotting as described above.
Results and Discussion
The construction of pSB1C3-Leish shuttle vector
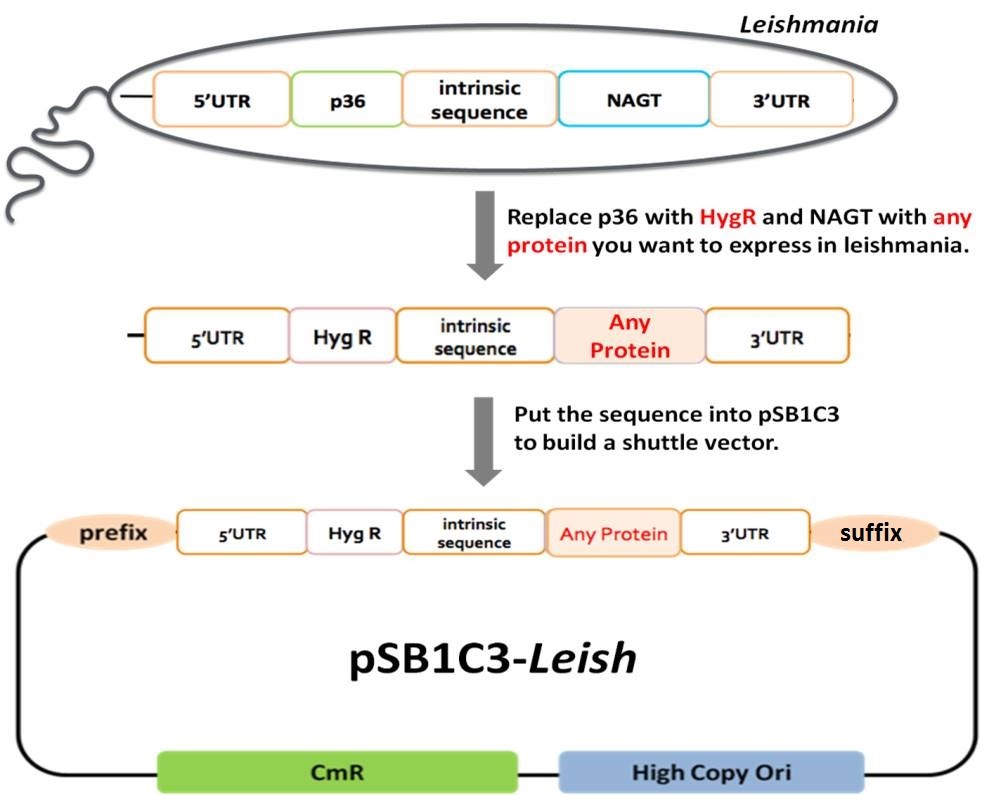
In order to use photo-inactivated Leishmania as a safe carrier to deliver specific antigens to the APCs for T cell stimulation, we designed an E. coli-Leishmania shuttle vector for antigen expression in Leishmania. Our design followed the biobrick standards to provide a standardized shuttle vector for our own experiment and for others’ future applications.
In Leishmania genome, p36 and nagt genes are separated by an intergenic region of about 2300 bp (Fig. 1). The first coding region p36 was replaced with hygromycin resistance (Hyg R) gene in the shuttle vector as a selection marker in Leishmania. The 5’UTR may contain promoter and ribosome binding site and other important functions in Leishmania. Therefore, 5’UTR was included as a biobrick part. The second coding region NAGT was designed to be replaced in the shuttle vector with any protein targeted for Leishmania expression. In our project, we planned to put OVA and H1N1 HA cDNAs into our pSB1C3-Leish shuttle vector. Therefore, we also created OVA and HA biobrick parts. The 3’UTR of the sequence was designed to function as a terminator part. The 2300-bp intrinsic sequence was also included in our biobrick design to regulate the expression of foreign proteins in the shuttle vector. pSB1C3 is a high copy number plasmid carrying chloramphenicol resistance (CmR) gene and can be expressed in E. coli. Therefore, we chose pSB1C3 as the backbone vector to build pSB1C3-Leish as the E. coli-Leishmania shuttle vector (Fig. 1).
The 2300-bp intergenic region could not be synthesized due to very high GC content. Our alternative approach was to separate the sequence into 3 parts. The plan was to amplify these 3 parts from p6.5 plasmid using PCR, and then ligated to construct the pSB1C3-2300intron part. Although the parts for 2300-bp intergenic region were amplified by PCR, the amplicon of final ligation product was only about 2-2.1 kb in length (Fig. 2, lane E). We repeated the experiment and still were unable to get the correct ligation product. As a result, we decided to remove the 2300-bp intergenic region from our original design. Instead, we generated two alternative constructs: pSB1C3-5’HYG-HA-3’UTR and pSB1C3-5’HYG-OVA-3’UTR. We planned to use these constructs to test the role of the 2300-bp intergenic region in protein expression from E. coli-Leishmania shuttle vector.
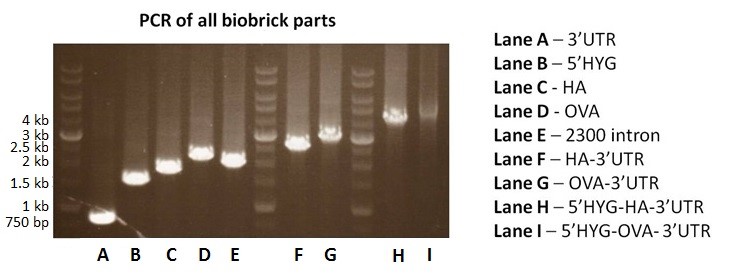
pSB1C3-HA-3’UTR and pSB1C3-OVA-3’UTR were constructed by ligating pSB1C3-3’UTR with pSB1C3-HA and pSB1C3-OVA, respectively. PCR analysis showed the correct amplicon size of 2.6 kb for HA-3’UTR (Fig. 2, lane F) and 2.9 kb for OVA-3’UTR (Fig. 2, lane G). They were subsequently ligated with pSB1C3-5’HYG to make pSB1C3-5’HYG-HA-3’UTR (BBa_K1955005) and pSB1C3-5’HYG-OVA-3’UTR (BBa_K1955006), respectively. PCR confirmed the construction of pSB1C3-5’HYG-HA-3’UTR and pSB1C3-5’HYG-OVA-3’UTR. The correct amplicon size was 4.1 kb for 5’HYG-HA-3’UTR (Fig. 2, lane H) and 4.5 kb for 5’HYG-OVA-3’UTR (Fig. 2, lane I).
Establishment of stable Leishmania transfectants
Our next goal was to determine whether pSB1C3-5’HYG-HA-3’UTR and pSB1C3-5’HYG-OVA-3’UTR could be successfully expressed in Leishmania. We used electroporation to transfect our constructs into Leishmania and tested the expression of the encoded proteins by Western blot assay. As a control to verify the accuracy of OVA cDNA, we put our pSB1C3-OVA part into pX63-HYG vector, a common vector known to express foreign proteins in Leishmania24. Both pSB1C3-5’HYG-OVA-3’UTR and pX63-HYG-OVA were transfected into Leishmania 12-DT strain and stable transfectants were selected by hygromycin. As shown in Fig. 3, OVA expression in Leishmania transfectants was examined by immunoblotting. Compared to the recombinant OVA protein (Lane 1, lower panel), neither Leishmania transfected with pSB1C3-5’HYG-OVA-3’UTR nor pX63-HYG-OVA expressed OVA protein (Lanes 3 and 4, lower panel). As expected, untransfected Leishmania showed no OVA expression (Lane 2, lower panel). The tubulin Western blot confirmed equal loading of Leishmania proteins (Lanes 2-4, upper panel).
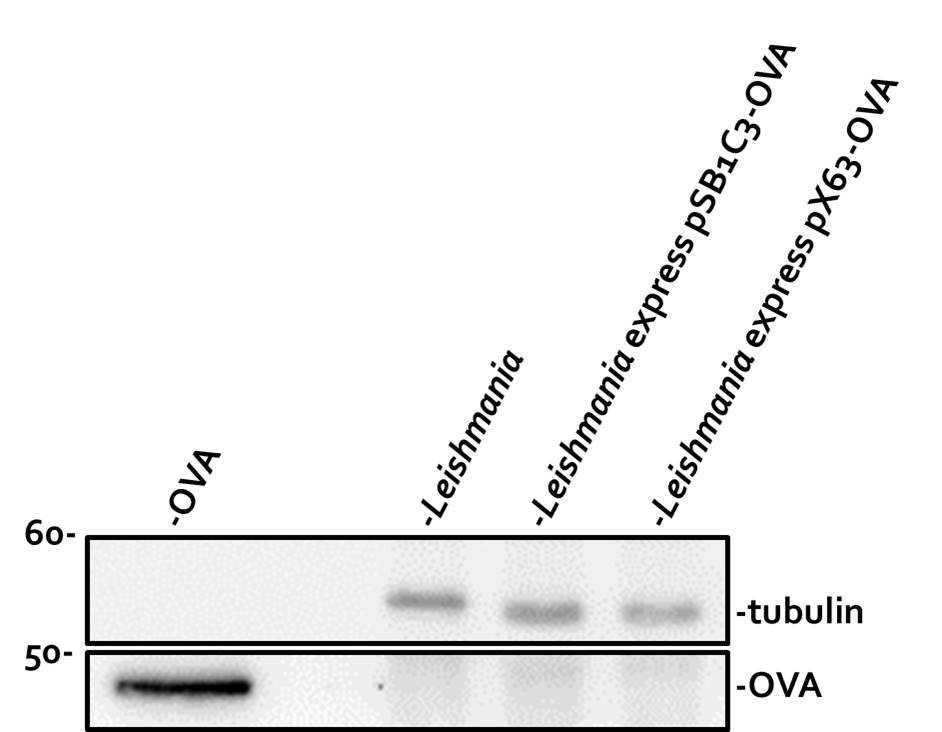
The observation that pX63-HYG-OVA could not express OVA protein in Leishmania strongly suggested that our pSB1C3-OVA part might have sequence errors. However, we could not rule out the possibility that the absence of 2300-bp intergenic region might also contribute to the loss of OVA expression from pSB1C3-5’HYG-OVA-3’UTR.
Next, we tested the expression of HA from pSB1C3-5’HYG-HA-3’UTR by immunoblotting (Fig. 4, Lanes 3 and 4). As shown in the top panel, no HA was detected in untransfected Leishmania (Lane 3) and the stable transfectant (Lane 4). Immunoblotting of Leishmania endogenous p36 showed more protein loading in untransfected Leishmania than in HA transfectant (compare Lanes 3 and 4, bottom panel). As a positive control for HA immunoblotting, we also included 293T cells without and with HA expression by lentiviral transduction (Fig. 4, Lanes 1 and 2). The result confirmed the ability of immunoblotting in detecting HA (top panel). Actin immunoblotting further verified equal loading of 293T proteins (middle panel).

The results from Fig. 3 and Fig. 4 suggested that the 2300-bp intergenic region was indeed essential for target protein expression from the shuttle vector. However, we could not exclude the possibility that, similar to pSB1C3-OVA, pSB1C3-HA might have sequence errors as well.
HA expression in E. coli
To check the accuracy of pSB1C3-HA, we used BL21 competent cells transformed with pSB1C3-HA to detect HA expression. We chose BBa_J04500 as a promoter to drive protein expression. BBa_J04500 has a LacI inducible promoter with RBS and can be induced by IPTG to activate transcription. Therefore, we built pSB1C3-J04500-HA and transformed it into E. coli BL21. Two independent clones #1 and #2 were isolated (Fig. 5, Lanes 3-6). As a positive control, we built pSB1C3-J04500-MAL-p5x-HA plasmid known to express maltose binding protein-hemagglutinin (MBP-HA) fusion protein (Fig. 5, Lanes 1 and 2). As a negative control, pUC19 empty vector was transformed into E. coli (Fig. 5, Lanes 7 and 8). All four clones were either left untreated (Lanes 2, 4, 6 and 8) or treated with IPTG (Lanes 1, 3, 5 and 7).

As shown in the upper panel (Lane 1), IPTG successfully induced the expression of MBP-HA fusion protein (approximately 104.76 kD). Similarly, IPTG induced HA expression from both clones of pSB1C3-J04500-HA (Lanes 3 and 5, lower panel). No signal was detected in the negative control (Lanes 7 and 8). This result confirmed the accuracy of our pSB1C3-HA part. It further supported the importance of 2300-bp intergenic region in correct protein expression from the E. coli-Leishmania shuttle vector.
Conclusions
We designed an E. coli-Leishmania shuttle vector to express foreign proteins in Leishmania. We were unable to put the 2300-bp intergenic region into the shuttle vector. Isolation of hygromycin-resistant Leishmania indicated successful expression of exogenous gene downstream of 5’UTR. The absence of OVA and HA protein expression from stable transfectants, on the other hand, strongly supported the essential role of the 2300-bp intergenic region in driving the downstream gene expression. The synthesized OVA cDNA might also have some unexpected sequence errors. For the future work, we will use other strategies to put the 2300-bp intergenic region into the shuttle vector to facilitate expression of foreign proteins. After confirming successful expression of foreign antigens in Leishmania, we will conduct in vivo test to fully investigate its safety and effectiveness as an adjuvant.
Acknowledgements
We thank all members of the Department of Biomedical Sciences at Chang Gung University and the Department of Molecular Parasitology and Tropical Diseases at Taipei Medical University for their support. In particular, we want to thank Prof. Shin-Ru Shih, Prof. Jin-Chung Chen, and Prof. Ming-Ling Kuo for their extensive support during our participation in iGEM. Moreover, we would like to thank all external sponsors, including GeneDireX, Body Organ Biomedical Corporation (BOBC), Integrated Device Technology (IDT), and Taqkey Science, for their support.
Response to Reviewers
A transcript of the reviewer comments and author responses from the Live Peer Review Jamboree can be found here: CGU-Taiwan Response to Reviewers
References
- Chan M. (2014). The contribution of immunization: saving millions of lives, and more. Public Health Reports 129 Suppl 3: 7-8.
- Centers for Disease Control and Prevention (1999). Impact of vaccines universally recommended for children – United States, 1990-1998. MMWR: Morbidity and Mortality Weekly Report 48(12): 243-248.
- Calugar A., Ortega-Sanchez I. R., et al. (2006). Nosocomial pertussis: costs of an outbreak and benefits of vaccinating health care workers. Clinical Infectious Diseases 42(7): 981-988.
- Roush S. W. and Murphy T. V. (2007). Historical comparisons of morbidity and mortality for vaccine-preventable diseases in the United States. JAMA 298(18): 2155-2163.
- Pierce S. K. and Miller L. H. (2009). World Malaria Day 2009: what malaria knows about the immune system that immunologists still do not. Journal of Immunology 182(9): 5171-5177.
- Bergmann-Leitner E. S. and Leitner W. W. (2014). Adjuvants in the driver’s seat: how magnitude, type, fine specificity and longevity of immune responses are driven by distinct classes of immune potentiators. Vaccines (Basel) 2(2): 252-296.
- Koup R. A. and Douek D. C. (2011). Vaccine design for CD8 T lymphocyte responses. Cold Spring Harbor Perspectives in Medicine 1(1): a007252.
- Rosendahl Huber S., Van Beek J., et al. (2014). T cell responses to viral infections – opportunities for peptide vaccination. Frontiers in Immunology 5: 171.
- Macleod M. K., Kappler J. W., et al. (2010). Memory CD4 T cells: generation, reactivation and re-assignment. Immunology 130(1): 10-15.
- Gilbert S. C. (2012). T-cell-inducing vaccines – what’s the future. Immunology 135(1): 19-26.
- Ahlers J. D. and Belyakov I. M. (2010). Memories that last forever: strategies for optimizing vaccine T-cell memory. Blood 115(9): 1678-1689.
- Wack A., Baudner B. C., et al. (2008). Combination adjuvants for the induction of potent, long-lasting antibody and T-cell responses to influenza vaccine in mice. Vaccine 26(4): 552-561.
- Coffman R. L., Sher A., et al. (2010). Vaccine adjuvants: putting innate immunity to work. Immunity 33(4): 492-503.
- De Donato S., Granoff D., et al. (1999). Safety and immunogenicity of MF59-adjuvanted influenza vaccine in the elderly. Vaccine 17(23-24): 3094-3101.
- Podda, A., and G. Del Giudice (2006). MF59: a safe and potent adjuvant for human use. Immunopotentiators in Modern Vaccines. V. E. Schijns, and D. T. O’Hagan, eds. Academic Press, Oxford, p. 149-159.
- Gupta R. K. and Siber G. R. (1995). Adjuvants for human vaccines – current status, problems and future prospects. Vaccine 13(14): 1263-1276.
- Gupta R. K., Relyveld E. H., et al. (1993). Adjuvants – a balance between toxicity and adjuvanticity. Vaccine 11(3): 293-306.
- Titus R. G. and Ribeiro J. M. (1988). Salivary gland lysates from the sand fly Lutzomyia longipalpis enhance Leishmania Science 239(4845): 1306-1308.
- Chang K. P. (2001). Leishmaniases. eLS, John Wiley & Sons, Ltd.
- Benov L. (2015). Photodynamic therapy: current status and future directions. Medical Principles and Practice 24 Suppl 1: 14-28.
- Sah J. F., Ito H., et al. (2002). Genetic rescue of Leishmania deficiency in porphyrin biosynthesis creates mutants suitable for analysis of cellular events in uroporphyria and for photodynamic therapy. Journal of Biological Chemistry 277(17): 14902-14909.
- Chang K. P. and Kolli B. K. (2016). New “light” for one-world approach toward safe and effective control of animal diseases and insect vectors from leishmaniac perspectives. Parasit Vectors 9(1): 396-409.
- Dutta S., Kolli B. K., et al. (2008). Transgenic Leishmania model for delta-aminolevulinate-inducible monospecific uroporphyria: cytolytic phototoxicity initiated by singlet oxygen-mediated inactivation of proteins and its ablation by endosomal mobilization of cytosolic uroporphyrin. Eukaryot Cell 7(7): 1146-1157.
- Dutta S., Ongarora B. G., et al. (2011). Intracellular targeting specificity of novel phthalocyanines assessed in a host-parasite model for developing potential photodynamic medicine. PloS One 6(6): e20786.