iGEM REPORT: Engineering E. coli for phosphate bioremediation
Note: This iGEM Report was submitted to the PLOS iGEM Realtime Peer Review Jamboree, and has not undergone formal peer review by any of the PLOS journals. We welcome your comments on this work.
Engineering E. coli for phosphate bioremediation
Suraj Mohan (1), Caleigh Roleck (2), Paige Rudin (3), Bowman Clark (2), Emma Foster (1), Archana Kikla (2), Sean Magill (1), Mark Aronson (1), Ryan Budde (3), Hana Kubo (2), Arren Liu (2), Helena Lysandrou (2), Casey Martin (1), Alexa Petrucciani (1), James Welch (2), Soo Ha (1), Jenna Rickus (1,3), Kevin Solomon (1)
- School of Agricultural and Biological Engineering, Purdue University
- Department of Biological Sciences, Purdue University
- School of Biomedical Engineering, Purdue University
*Corresponding author: [email protected]
Author Contributions
Conceptualization: MA RB BC EF AK HK AL HL CM SMa SMo AP CR PR JW
Data curation: BC EF SMo CR PR
Formal analysis: BC CR
Funding acquisition: AL SMa SMo PR JW
Investigation: BC EF SMa SMo CR PR
Methodology: BC MA EF CR PR
Project administration: BC PR
Resource: SH JR KS
Supervision: SH JR KS BC MA RB HK AL SMo CM JW
Visualization: AL CR PR
Writing – Original Draft Preparation: SMo AK CR PR
Writing – Review and Editing: RB HK AL HL CM SMo AK JR CR PR KS JW
Abstract
Lakes and coastal regions are increasingly threatened by harmful algal blooms driven by high concentrations of phosphorus, often from domestic and agricultural fertilizers. Algal blooms decrease water quality, interrupt the function of critical infrastructure, and harm businesses reliant on affected bodies of water, disturbing both the environment and the economy. Still, despite the damage that excess phosphorus can cause, phosphorus is a limited resource and a vital nutrient required for agriculture. To improve phosphate management, we plan to develop a strain of modified Escherichia coli that can accumulate and store large amounts of phosphorus, while also being able to release stored phosphorus in a controlled manner. As E. coli is both robust and easy to engineer, a phosphorus management system utilizing E. coli can be used in a wide range of environmental conditions and can be adapted to meet the specific needs of each application scenario and environment. To create our system, we identified genes putatively responsible for phosphate uptake, storage, and preparation for exportation in the polyphosphate-accumulating organism Microlunatus phosphovorus. We then transformed these genes into E. coli to characterize functions of these previously uncharacterized proteins. Concurrently, we built a bioreactor and designed a suite of cost-effective phosphorus reclamation modules around xerogel-immobilized cells for contained, multipoint phosphate bioremediation. Xerogel beads are a porous glass matrix which entrap cells but allow water, phosphates, and other nutrients to flow through. Characterization of the exopolyphosphatase (PPX2) homolog revealed that PPX2 leads to increased phosphorus release, and preliminary characterization of the polyphosphate kinase 2 (PPK2) homolog C suggests that PPK2 homolog C is potentially responsible for polyphosphate hydrolysis. Through applied genetic, chemical, and mechanical engineering principles we expect to provide a means for preventing harmful algal blooms in both developed and developing countries while also recovering phosphorus for later agricultural use.
Financial Disclosure
Funding was generously offered by Purdue University, Agricultural and Biological Engineering Department, College of Agriculture, College of Science, Honors College, Office of the Executive Vice President of Research, College of Engineering, Learning Beyond the Classroom Grants, Day of Giving Donations, and Molecular Agriculture Summer Institute stipends. Non university sources include SYNENERGENE, Experiment Crowdfunding Income, and Monsanto. Synthetic DNA was donated by Integrated DNA Technologies. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
Partial funding received from external commercial sources such as Monsanto and Dow AgroSciences, did not affect project design or execution. Donations and grants were used to purchase laboratory equipment and supplies for protocols planned prior to funding acquisition.
Data Availability
Yes- all data are fully available without restriction. Data can be accessed at https://osf.io/snrgg/.
Introduction
Water phosphates in excess of 25 micrograms per liter are known to drive growth of harmful algal blooms (HABs) during temperatures warmer than 25°C. These blooms compromise water quality by choking oxygen from aquatic ecosystems, and leaching neurotoxins and hepatotoxins into sources of potable water. In doing so, HABs cost global industry more than ten billion USD in damage and threaten human health every year. In addition, there are currently no federal restrictions in the US on water phosphate pollution. Phosphorus is a nonrenewable resource, and in the near future, supply will fail to meet global demands. Therefore, better recycling or reclamation methods must be established (1).
In order to remove excess phosphorus from water sources, a three part system in Escherichia coli is envisioned: 1) capture phosphorus from water, 2) store phosphorus as polyphosphate, and 3) release phosphorus from cells on demand. Bacteria are embedded in engineered xerogel beads to prevent their release into the wild and take up luxury amounts of phosphorus, a system adapted from phosphorus accumulating organisms (PAOs), which are currently used in waste treatment plants. These PAOs generate energy by storing phosphate as polyphosphate. ATP dependent polyphosphate kinases bind phosphate into polyphosphate polymers for storage. The genes that normally cleave phosphates from this chain are downregulated until expressed by a user-defined trigger. The cells then release much of their phosphate and deposit it into the surrounding media. The media can be transported elsewhere while the cells stay embedded in the xerogel beads. This phosphate-enriched media can be further refined to add value to the product. The system offers a wide range of applications including methane digesters, tile drainage systems, wastewater treatment plants, home bioreactors, and floating phosphorus collector units.
Microlunatus phosphovorus is a gram-positive, aerobic, coccus-shaped, actinobacteria of the relatively new genus Microlunatus. First isolated in 1995 from activated sludge in the wastewater treatment process, M. phosphovorus demonstrates remarkable phosphorus removal from wastewater by being able to take up to 48% dry weight in phosphorus alone (2). After Kawakoshi et al. sequenced M. phosphovorus’s genome, phylogenetic analysis revealed the presence of genes that putatively coded for proteins regulating the organism’s phosphorus management system: four polyphosphate kinases (PPK), two polyphosphate-dependent glucokinases (PPGK), three phosphate transporters (PiT), and one exopolyphosphatase (PPX) (3) (Fig. 1).

Polyphosphate kinases catalyze the transfer of polyphosphates between nucleotide mono-, di-, or tri-phosphates and polyphosphate chains, thus leading to the synthesis or hydrolysis of polyphosphate chains. Of the four putative PPKs, one belongs to the subtype PPK1, which favors polyphosphate synthesis using ATP. Two of the putative PPKs belong to the subtype PPK2, which have varying activities for polyphosphate synthesis and hydrolysis. Based on phylogenetic analysis, PPK2 homolog A likely favors polyphosphate synthesis and PPK2 homolog B likely favors polyphosphate hydrolysis. The third PPK, which will be referred to as PPK2 homolog C, has been placed in a phylogenetic cluster consisting of PPKs with underdetermined function, although this PPK known as PPK2 homolog C has similarities to both a PPK2 and a polyP-dependent AMP phosphotransferase (PAP), a third PPK subcluster that favors polyphosphate hydrolysis (3).
Polyphosphate glucokinases phosphorylate glucose to glucose-6-phosphate using available polyphosphate or ATP. Of the two PPGK homologs present in M. phosphovorus, one has been previously characterized; a team from Hiroshima University found that this PPGK can only phosphorylate glucose with polyphosphate and cannot use ATP, (2), thus this homolog will be referred to as PPGK ATP-independent (PPGK ATPI). It is unknown whether the second PPGK homolog is able to use ATP. The inorganic phosphate transporters (Pit) actively transfer inorganic phosphorus in and out of the cell using a proton gradient. These PPK, PPGK, and Pit genes putatively play critical roles in polyphosphate accumulation in M. phosphovorus by aiding in the phosphate transport and polyphosphate construction. M. phosphovorus also seems to only express one exopolyphosphatase (ppx) gene rather than the two that are typically observed in other Actinobacteria (3). PPX can be utilized for its ability to hydrolyze terminal phosphates of polyphosphate chains in order to minimize the overall amount of orthophosphate leaving from the system. (Fig. 1.) While this organism holds significant promise for application in enhanced biological phosphorus removal (EBPR), not enough characterization has been done with it to engineer an optimized, robust PAO.
In order for the implementation of a synthetic biology solution to be feasible, an effective system of containment to prevent organisms from escaping into the environment must be developed. For example, if microorganisms capable of accumulating large quantities of phosphorus were to grow unchecked in an ecosystem, plants and animals may perish from lack of this essential nutrient. In the context of this project, modified E. coli were incorporated into xerogel silica beads created using the sol-gel method. These beads, several millimeters in diameter, encapsulated microorganisms within a porous matrix that allowed water, phosphorus, and other nutrients to flow through but immobilized bacteria, effectively containing them and restricting their growth.
The combination of biological construct and physical mechanism compose a novel system that could effectively remove phosphorus from water, store it for later use, and recycle this nutrient by releasing it on demand. Through assembling a genetic circuit using standard synthetic biology procedures and iGEM materials and protocols, measuring the efficacy of the modified organism, and creating structures to contain and support the system, a solution to the global rise in the growth of HABs is proposed.
Materials and Methods
Plasmid assembly and transformations
We ordered all genes as gBlocks (Integrated DNA Technologies, Coralville, IA), using the GenBank sequence for each of the ten genes except PPK2 homolog C, for which we used the NCBI Reference Sequence. (Table 1). The sequences for each gene, as well as their putative function, was determined by Kawakoshi et al in 2012 (3).
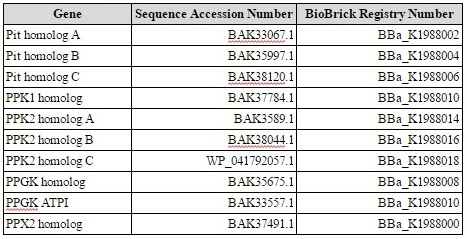
With the exception of PPK1 homolog, we ordered all genes as a single gBlock. All gBlock segments began with a HindIII restriction site at the 3ʹ end, followed with the weak, constitutive Anderson promoter BBa_J23015, the standard BioBrick prefix, containing an EcoRI and an XbaI restriction site, and the ribosome binding site BBa_J34803. The 10X polyhistidine tag BBa_K844000 was fused to the c-terminus of every protein, with the exception of PPK2 homolog A, which had a 6X polyhistidine tag fused to the n-terminus due to synthesizing complexities. We split the two gBlocks for PPK1 homolog in the middle of the gene. Following each gene, the gBlocks had the standard BioBrick suffix, containing a SpeI and Pst1 restriction site, the terminator BBa_B0010, and then a HindIII restriction site at the 5ʹ end.
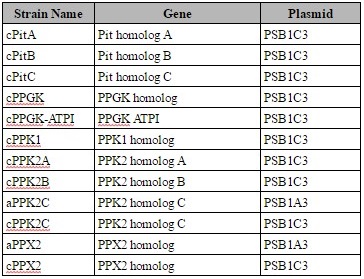
After we received the gBlocks, we amplified them via PCR. We designed the forward primer (Thermo Fisher Scientific, Waltham, MA) using the sequence for the HindIII restriction site and the first 18 bases of the Anderson promoter and adding the bases “ctat” prior to the HindIII restriction site (ctataagctttttacggctagctcagtc). We designed the reverse primer (Thermo Fisher Scientific, Waltham, MA) using the complementary sequence for the HindIII restriction site and the last 14 bases of the terminator and adding the bases “atat” prior to the restriction site (atataagcttgagagcgttcaccg). Gibson assembly was used to join together the two PPK1 homolog gBlocks, as described in Nature Methods (4). Then, we performed 3A assembly to ligate each gene and the medium strength constitutive Anderson promoter BBa_J23016 into the plasmid PSB1C3 or PSB1A3. 3A assembly was performed using the standard iGEM protocol (5). We transformed the plasmid into NEB 5-alpha Competent E. coli in accordance with NEB’s recommended protocol (6) and plated the transformants on LB agar plates with either chloramphenicol (for PSB1C3 plasmids) or ampicillin (for PSB1A3 plasmids), producing twelve different strains (Table 2). Colonies were inoculated and mini-prepped. We submitted the plasmids to the Purdue Genomics Core Facility to validate the sequence via Sanger sequencing.
Analysis of phosphorus uptake, storage, and release
We inoculated strains of E. coli containing genetic constructs theorized to lead to increased phosphorus uptake or increased phosphorus exportation in LB broth. The culture was then diluted to produce 250 mL of a minimal medium culture with an optical density of 0.01 at 600 nm. After inoculation, we took 5 mL samples hourly for 6 hours, starting with hour 0. We froze all samples at −20 °C for later analysis. We collected an additional sample at 72 hours. After sample collection, we centrifuged the samples to separate the cellular component from the supernatant and transferred the supernatant to a second tube. The total phosphorus content of the supernatant was quantified using an Inductively Coupled Plasma (ICP) spectrometer from the United States Department of Agriculture National Soil Erosion Research Lab.
In addition, we inoculated all strains in 5 mL of LB broth supplemented with K2HPO4 to produce a 2 mM PO43- solution, which is deemed a sufficient media concentration to assess phosphorus accumulation (7). We then extracted polyphosphate granules and quantified polyphosphate concentration spectrophotometrically with a toluidine blue and acetic acid solution, in accordance with the Mukherjee and Ray protocol (8). The absorbance of each sample at 580 nm was compared against a standard curve prepared using sodium phosphate glass type 45.
Production of silica beads
We formed silica beads as a means of immobilizing bacteria. The sol-gel process combines an acidic hydrolyzed solution of tetramethyl orthosilicate (TMOS) with E. coli suspended in neutral phosphate-buffered saline (PBS), as described in Rickus et al (8). When the TMOS sol-precursor is added to the buffer, the pH increase favors rapid polymerization of sol-gel at room temperature. To form the silica beads, we dripped the TMOS sol-precursor and PBS mixture into a vat of mineral oil using a syringe.
To verify that the silica beads can effectively immobilize E.coli, we produced a red fluorescent protein-expressing strain of E. coli using the BioBrick construct BBa_J04450 (a LacI regulated promoter, ribozyme binding site, an engineered Discosoma striata red fluorescent protein mutant, and a double terminator) in the plasmid PSB1C3. We then added this strain to the PBS, which was used in the production of silica beads. The beads were then imaged with the EVOS Cell Imaging System in order to detect red fluorescence both within and between the beads.
Bioreactor construction and iterative design
Seeking a solution that could be implemented in a variety of different scenarios, we consulted public and private stakeholders to determine a list of design specifications with quantitative metrics for a desirable physical prototype, such as a cost of less than $100 USD to construct, $25 to maintain annually, and a water phosphorus concentration less than 0.025 ppm post-filtration. In collaboration with the United States Department of Agriculture National Soil Erosion Research Laboratory (USDA-NSERL), we constructed a bioreactor to allow water to flow over and through silica beads, emerging with a lesser concentration of phosphorus after passing through the system. A central goal of this design was to produce a prototype that could, with a few modifications, be practical in many environments, including agricultural field tile drains, within wastewater treatment facilities, in septic tank water treatment systems, and below city streets in sewer drains.
Two five-gallon buckets–one “reactor” and one “reservoir”–are connected by one-inch flexible tubing and an aquarium pump powered by an electrical outlet to keep the water level stable within the reactor bucket (Fig. 2). Three exit ports around the base of the reactor bucket allow gravity-driven effluent to flow through an additional length of tubing into one of three water filter canisters. Each canister, or phosphorus reclamation unit, contains an inner water filter; between the water filter and outer canister wall, silica beads containing modified microorganisms are packed. Water to be treated would enter the reservoir bucket, be pumped into the reactor bucket, exit the reactor bucket through its base, then flow through a filter canister and bead matrix where its phosphorus concentration is reduced before leaving the system via the outflow spigot (Fig. 2).
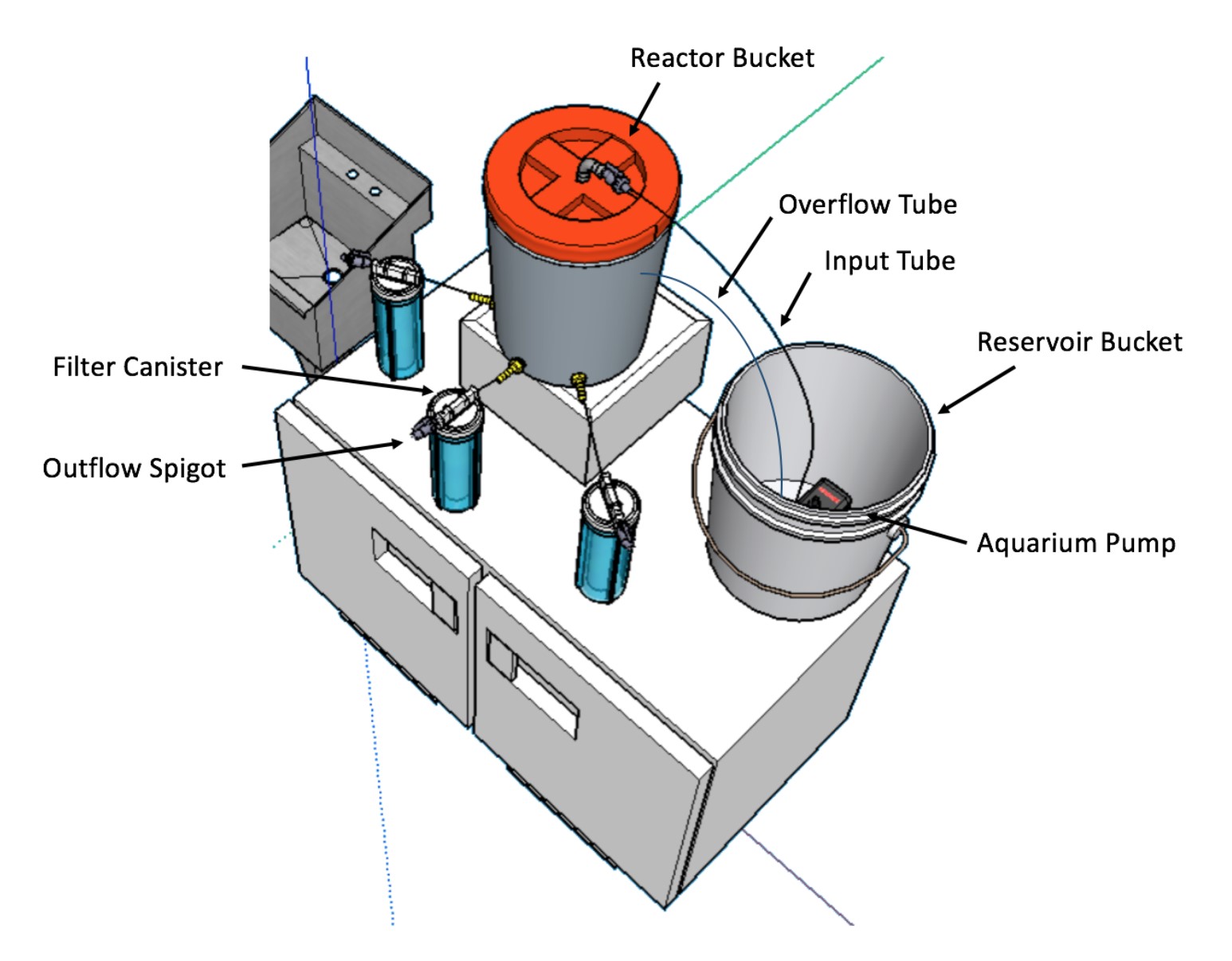
A theoretical design iteration includes the replacement of the filter canisters with a bead-packed pipe. This pipe would decrease water velocity as its diameter decreases, enabling maximum phosphorus uptake by bacteria within the beads. Diameter could be adjusted to optimize the volume of water capable of passing through the system and the amount of phosphorus that could be absorbed from it.
Results and Discussion
Plasmid assembly and transformations
Sequencing results confirmed that only the transformants for the strains aPPK2C, aPPX2, and cPPX2 contained the correct insert. Sequencing results for all other strains indicated that colonies only contained the PSB1C3 plasmid without the correct insert, and so the plasmid self-ligated. In future experimentation, we plan to treat the plasmid with phosphatase to remove the 5ʹ phosphate and prevent self-ligation of the backbone when assembling the plasmids for the other strains.
Analysis of phosphorus uptake, storage, and release
We inoculated the strain aPPX2 and an unmodified strain of E. coli in minimal media and allowed to grow for 72 hours. After centrifuging the samples and analyzing the total phosphorus content of the supernatant with an Inductively Coupled Plasma spectrometer, we compared the total phosphorus concentration on both supernatants. We expected the phosphorus concentration of aPPX2’s supernatant to be greater than that of unmodified E. coli, as PPX2 cleaves the terminal phosphate from a polyphosphate chain, thus preparing an orthophosphate molecule for exportation. Therefore, since aPPX2 contains a putative PPX2, and the unmodified strain does not, aPPX2 should theoretically release more phosphorus into the supernatant than unmodified E. coli. The concentrations of total phosphorus in the supernatant were determined to be 2.412 ppm and 0.741 ppm, respectively, and so, as expected, the supernatant for aPPX2 had a greater total phosphorus than that of unmodified E. coli and the PPX2 homolog is behaving as expected. (Fig. 3). A t-test indicated that PPX2-producing E. coli had significantly more (p < 0.005) phosphorus in the supernatant than that of unmodified E. coli. The other sequence verified strains, aPPK2C and cPPX2, were not analyzed in this manner, as cPPX2 contains the same gene as aPPX2, and aPPK2 contains the gene for a putative polyphosphate kinase, which would not produce a change in phosphorus uptake or exportation, only polyphosphate storage.

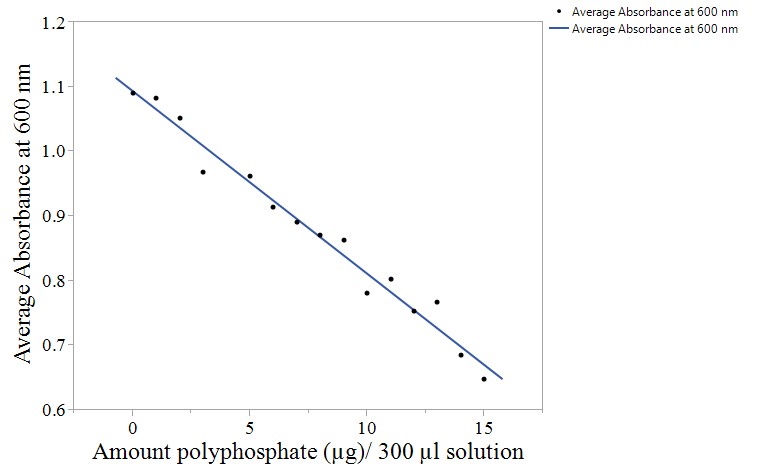
We used a standard curve to estimate the intracellular polyphosphate concentration of several strains of E. coli. The standard curve compares absorbance at 630 nm to polyphosphate concentration, and had an R2 value of 0.975 and a correlation of −0.987 (Fig. 4). As this is a strong correlation, a linear model was concluded to be a reasonable way to estimate intracellular polyphosphate concentration colorimetrically.
The amount of intracellular polyphosphate was then estimated for four strains of E. coli: aPPK2C, aPPX2, cPPX2, and an unmodified strain. We hypothesize that both aPPX2 and cPPX2 should have less intracellular polyphosphate than unmodified E. coli, as both contain a putative PPX2, which hydrolyzes polyphosphate. Given the current knowledge on aPPK2C, we hypothesize that the intracellular polyphosphate concentration for aPPK2C would be different than that of unmodified E. coli, but we cannot predict whether the concentration would be higher or lower, as PPK2 homolog C’s activity for polyphosphate synthesis or hydrolysis could not be predicted based on phylogenetic analysis (3). All three modified strains were estimated to have a lower concentration of intracellular polyphosphate in comparison with unmodified E. coli (Fig. 5). Still, pairwise t-tests did not detect any statistical significance with an α-level of 0.05. When compared to the unmodified strain, aPPK2C had a p-value of 0.3969, aPPX2 had a p-value of 0.3029, and cPPX2 had a p-value of 0.5433. Still, as we only obtained data for three samples for each strain, the lack of statistical significance could potentially be due to low sample size.
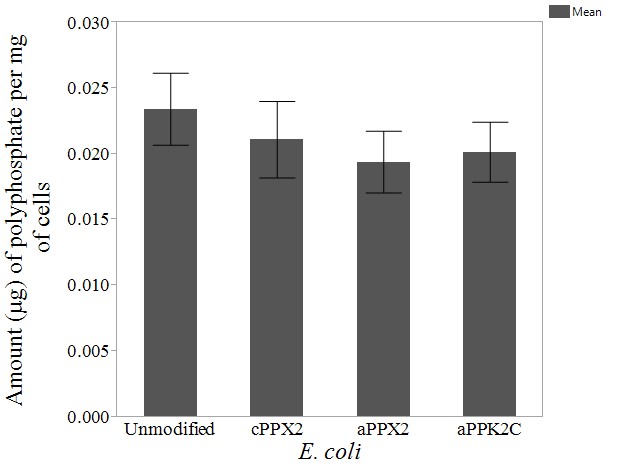
The results suggest the function of the putative PPX2 homolog as an exopolyphosphatase, as PPX2 homolog expression in E. coli increased phosphate release, and may lower intracellualr polyphosphate concentration in E. coli, although further testing is needing. This PPX2 homolog was previously uncharacterized in both M. phosphovorus and E. coli. Despite M. phosphovorus being gram-positive and E. coli being gram-negative, the PPX2 homolog should still produce a functional protein because PPX2 is not an integral membrane protein. While exopolyphosphatase leads to the expulsion of phosphorus from the cell, PPX2 does this by preparing intracellular phosphorus for expulsion through the hydrolysis of polyphosphate to produce orthophosphate. Therefore, differences in cell wall and membrane structure should not affect the ability of E. coli to produce a functional exopolyphosphatase native to E. coli.
Due to the lack of statistical significance for results for strains containing PPK2 homolog C, we cannot confirm the function of PPK2 homolog C, although results suggest that PPK2 homolog C expression may decrease the amount of intracellular polyphosphate. Further testing is needed. Still, despite the levels of uncertainty associated with these results, these results are the first indication of the activity for PPK2 homolog C for polyphosphate synthesis or hydrolysis. Not only was PPK2 homolog C previously uncharacterized in both M. phosphovorus and E. coli, but phylogenetic analysis placed PPK2 homolog C in a distinct cluster of putative PPK2 homologs with undetermined functions. Still, while PPK2 homolog C belongs to this distinct cluster of polyphosphate kinases, PPK2 homolog C is similar to both a PPK2 and a polyP-dependent AMP phosphotransferase (PAP), another polyphosphate kinase subtype. While the PPK2 polyphosphate kinase subfamily contains PPKs with activities as either a polyphosphate synthase or hydrolase, PAPs have an affinity for polyphosphate hydrolysis (3). There, these preliminary results and PPK2 homolog C’s similarity to a PAP as well as a PPK2 have caused us to form the hypothesis that PPK2 homolog C has polyphosphate hydrolase activity. Once again, despite the differences in cell wall and membrane structure, E. coli should be able to produce a functional PPK from M. phosphovorus, as PPK, as PPK2 homolog C is not an integral membrane protein. M. phosphovorus does contain four PPK homologs though, and while we were interested in all homologs, we only studied PPK2 homolog C, as aPPK2C was the only strain containing a sequence-verified plasmid with a PPK homolog.
Production of silica beads
After producing a strain of E. coli expressing a red fluorescent protein, immobilizing the strain within silica beads, and producing silica beads using the same method, but without adding E. coli, we viewed both samples of silica beads with an EVOS Cell Imaging System to analyze red fluorescence. The beads containing red fluorescent protein (RFP)-expressing E. coli displayed red fluorescence, while the negative control beads that did not contain E. coli did not exhibit red fluorescence. (Fig. 6.) As the red fluorescence was present within silica beads produced to contain red-fluorescent E. coli, these results indicate that the silica beads successfully immobilized the E. coli. If the beads do not contain immobilized E. coli, they would not exhibit fluorescence, just as the silica beads produced without E. coli did not exhibit fluorescence. Additionally, we did not detect red fluorescence between the beads for the sample of beads that contained RFP-expressing E. coli. This absence of fluorescence preliminarily indicates that the E. coli is not leaching out of the beads, as, if red-fluorescent E. coli leached out of the silica beads, red fluorescence outside of the beads would be detected. Still, in the future, more extensive leaching assays will be conducted. Additionally, while previous studies have shown that bacteria have 40% cell viability after one month of encapsulation in silica beads when glycerol is used as an additive to the silica beads (10), we plan on studying the viability of our own strain of polyphosphate-accumulating E. coli in the future.

If poor cell viability proves detrimental to implementation of our system, one possible mitigation strategy would be to vacuum evaporate the alcohol that results as a byproduct of the hydrolysis and polymerization reaction used to form the silica beads (11). Another mitigation strategy for poor viability would be to clump cells prior to encapsulation in the silica beads, as previous studies have concluded that this method improves cell viability (12). Additionally, the clumped cells would be less likely to leach out of the silica bead pores than the individual bacterium, as the radius of the clump would be larger. Still, if clumping the cells is insufficient or negatively affects implementation, significant cell leaching could also be addressed by engineering the E. coli to be dependent on an additive to the silica beads.
Conclusions
Due to the flexibility of this technology, future improvements and additional application scenarios are limited only by imagination. After adding an inducible promoter to the genetic construct within E. coli governing phosphorus exportation, the system would be able to store phosphorus within it to be released after the promoter is triggered. A kill switch could also be added to improve containment efficacy, decreasing the probability that no modified organism could disrupt the environment. Continued iterations of the bioreactor would enable further customization for different applications; for example, increasing the size of the reservoir bucket and the number of exit ports from the reactor bucket would increase the volume of water that could pass through the system. The addition of other genes such as those related to nitrogen uptake, pesticide digestion, and pharmaceutical breakdown would expand the number of pollutants capable of being captured and reclaimed by the bioreactor system. After adding these genes, the entire genetic construct could be integrated into the E. coli genome, making the organism a truly synthetic solution with immense positive societal and industrial impact.
Within the scope of a summer, genes from recently-sequenced M. phosphovorus genome were characterized individually, namely PPX2 homolog and PPK2 homolog C, and novel combinations imagined, revealing behaviors mirroring predictions given observed homologies. The complementary bucket bioreactor demonstrated its ability to effectively circulate water, indicating future diverse applications through continued iteration.
Acknowledgements
This work would not have been possible without the tremendous assistance of many. Teammate Barrett Davis furthered all steps of the experimental process, serving as an integral member of the resource team. Our advisors, Dr. Jenna Rickus, Dr. Kevin Solomon, and Dr. Kari Clase, offered continuous support, advice, and expertise. The bioreactor prototype and use of phosphate analytical equipment was generously facilitated by our partners at the USDA-NSERL–Dr. Ashley Hammac, Dr. Chi-hua Huang, Dr. Javier Gonzalez, Ms. Janae Bos, Ms. Brenda Hofmann, Mr. Stan Livingston, and Mr. Scott McAfee. The Wabash River Enhancement Corporation’s Ms. Sara Peel and Ms. Angela Andrews were instrumental in promoting our understanding of standard wastewater treatment practices and its impacts on our local community. Various departments and independent faculty members within Purdue in addition to industry partners Dow AgroSciences, SYNENERGENE, and Monsanto financially supported the endeavor. Thank you to all who made this summer a transformational experience for all involved; we truly could not have done it without you. Boiler up!
Response to Reviewers
A transcript of the reviewer comments and author responses from the Live Peer Review Jamboree can be found here: Purdue Response to Reviewers
References
- Kudela RM, Berdalet E, Bernard S, Burford M, Fernand L., Lu S, et al. Harmful algal blooms. A scientific summary for policy makers. Paris: IOC/UNESCO, 2015. 20 p. IOC/INF-1320.
- Tanaka S, Lee SO, Hamaoka K, Kato J, Takiguchi N, Nakamura K, et al. Strictly polyphosphate dependent glucokinase in a polyphosphate-accumulating bacterium, Microlunatus phosphovorus, Bacteriol. 2003 Sep; 185(18): 5654-5656.
- Kawakoshi A, Nakazawa H, Fukada J, Sasagawa M, Katano Y, Nakamura S et al. Deciphering the genome of polyphoshate accumulating actinobacterium Microlunatus phosphovorus. DNA Res. 2012 Oct; 19(5): 383-394.
- Gibson DG, Young L, Chuang R, Venter JC, Hutchison CA III, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods. 2009 Apr; 6: 343-345.
- International Genetically Engineered Machine. 3A Assembly [Internet]. Cambridge MA: iGEM; [cited 2017 Feb 18]. Available from http://parts.igem.org/Help:Assembly/3A_Assembly.
- New England Biolabs. High Efficiency Transformation Protocol (C2987H) [Internet]. Ipswich MA: NEB; 2014 [updated 2014 Oct 6; cited 2017 Feb 18]. Available from https://www.protocols.io/view/High-Efficiency-Transformation-Protocol-C2987H-imst3v.
- Morohoshi T, Maruo T, Shirai Y, Kato J, Ikeda T, Takiguchi N. Accumulation of inorganic polyphosphate in phoU mutants of Escherichia coli and Synechocystis Strain PCC6803. Appl Environ Microbiol, 2002 Aug; 68(8): 4107-4110. (2002)
- Mukherjee C, & Ray K.. An improved method for extraction and quantification of polyphosphate granules in microbial cells. Protoc Exch, 2015 Jul.
- Rickus J L, Chang PL, Tobin AJ, Zink JI. Dunn B. Photochemical coenzyme regeneration in enzymatically active optical material. J. Phys Chem, 2004 May; 108(26) 9325-9332
- Nassif N, Roux C, Coradin T, Rager MN, Bouvet OMM, Livage J. A sol-gel matrix to preserve the viability of encapsulated bacteria. J. Mater Chem, 2002 Dec; 13: 203-208.
- Ferrer ML, Yuste L, Rojo F, del Monte F. Biocompatible sol-gel route for encapsulation of living bacteria in organically modified silica matrixes. Chem. Mater, 2003 Aug; 15(19): 3614-3618.
- Nieto A, Areva S, Wilson T, Viitala R, Vallet-Regi M. Cell viability in wet silica gel. Acta Biomat, 2009 May; 5(9): 3478-3487.