iGEM Report: Integrated SynBio Tools Applied for Optimized Bioproduction of Poly-Lactic Acid
Note: This iGEM Report was submitted to the PLOS iGEM Realtime Peer Review Jamboree, and has not undergone formal peer review by any of the PLOS journals. We welcome your comments on this work.
Integrated SynBio Tools Applied for Optimized Bioproduction of Poly-Lactic Acid
A. Pandi (1), R. Ramírez-Garcia (1), J. Debédat (1), D. Dessaux (1), V. Gureghian (1), J. Hartunians (1), T. Mhoumadi (1), C. Nayrac (1), T. Ratovomanana (1), M. Saaidy (1), J. Tellechea (1), C. Jacry (1), A. Iglesias (1), M. Bargués-Ribera (1) *
- iGEM Évry-Genopole 2016, Institute of Systems and Synthetic Biology, Genopole, CNRS, Université d’Évry-Val-d’Essonne, Évry, France
* Corresponding author: M. Bargués-Ribera ([email protected])
Author Contributions
Conceptualization: AP MBR
Methodology: AP MBR
Investigation: AP RRG JD DD VG JH TM CN TR MS JT MBR
Writing – Original Draft: AP MBR RRG
Writing – Review & Editing: AP RRG JD DD JH TR MBR
Supervision: CJ AI
Abstract
In recent years, the advent of synthetic biology has enabled metabolic engineers to develop microorganisms as cell factories for bioproduction. Advanced engineering techniques have improved control of metabolic and genetic circuits, but new tools are still needed for optimal design of microorganisms. The aim of this report is to provide a systematic plan for facilitating the integration of rational engineering tools in biosynthesis processes. We define a methodology based on A) pathway enumeration; B) chassis choice; C) production optimization; and D) pathway implementation. A case study on the bioproduction of PLA, as performed on the Évry iGEM 2016 project, is presented as an example of design approach.
Financial Disclosure
Funding was provided by Genopole, Institute of Systems and Synthetic Biology, Université d’Évry-Val-d’Essonne, Integrated DNA Technologies, Laboratoire Informatique Biologie Intégrative et Systèmes Complexes (IBISC), French Embassy in the United States of America, CROUS Versailles, New England Biolabs, Geneious, MathWorks, Maison des Initiatives Étudiantes, and Grand Paris Sud.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
The authors have declared that no competing interests exist.
Ethics Statement
N/A
Data Availability
Yes – all data are fully available without restriction in our wiki: http://2016.igem.org/Team:Evry
Introduction
Initially described in the pages of Léduc and Loeb’s essays one century ago [1,2], synthetic biology has recently emerged as a promising subject on the boundary of diverse fields such as molecular biology, biotechnology and engineering. Its definition relies on the application of engineering principles to understand and modify life, and identifies the cell as a controllable entity with parts that are standardizable and modular [3,4].
Techniques of synthetic biology have become crucial for metabolic engineering of microorganisms by conceiving of them as machines or cell factories. In the last decade, omics technologies have contributed to an accurate description of gene regulatory systems of these microbial factories, as well as its metabolic pathways. Concerning this, synthetic biology tools have facilitated the study of their optimization and tuning, providing a new paradigm that analyzes all the elements and increases production efficiency [5-9].
Synthetic biology has brought forward engineering techniques that have improved robustness and control on metabolic and genetic circuits. These circuits, when described by mathematical models, have a calculable behavior and it is possible to predict the effect of particular components and mechanisms on the production flux distribution as well as dynamics [3,10,11]. Further, stoichiometric modelling of metabolic networks and dynamic simulation using a synthetic feedback regulation are promising approaches [12,13]. However, further efforts are needed to combined omics and synthetic biology tools for cellular design [14].
The goal of this report is to present the design methodology employed by the Team Évry during the iGEM competition in 2016, which can serve as a practical model to design optimized synthetic bioproduction of a compound of interest. In this methodology, a step by step procedure is presented, from choosing a host cell factory to searching, optimizing and finally implementing the pathway.
In the case study project, the initial objective was the bioproduction of Poly-Lactic Acid (PLA), a polymer used as bioplastic, and its further manufacturing and preparation for real-life applications. A design was described based on the literature that would enable further optimization compared to previous attempts of PLA bacterial production [15]. Due to several problems during the wet-lab experimental part and a lack of time to troubleshoot them, PLA was not obtained in the limit time of iGEM competition. However, positive feedback was received on the chosen chassis and modeling and optimizations was performed.
For this reason, this report organizes the steps followed for bioproduction optimization into an integrative methodology, using the PLA project as example case study; becoming a potential guide for future iGEM participants or synthetic metabolic engineers.
Methodology
Metabolic engineering is being informed by the synthetic biology framework of biological parts. Thus, our methodology provides a standard procedure that can be applied to manufacture a given product using the sustainable cell factory. Herein, we present a step by step methodology to follow, once the compound that one attempts to produce is known, from choosing the host chassis to optimizing and implementing the designed system, as resumed in Figure 1.
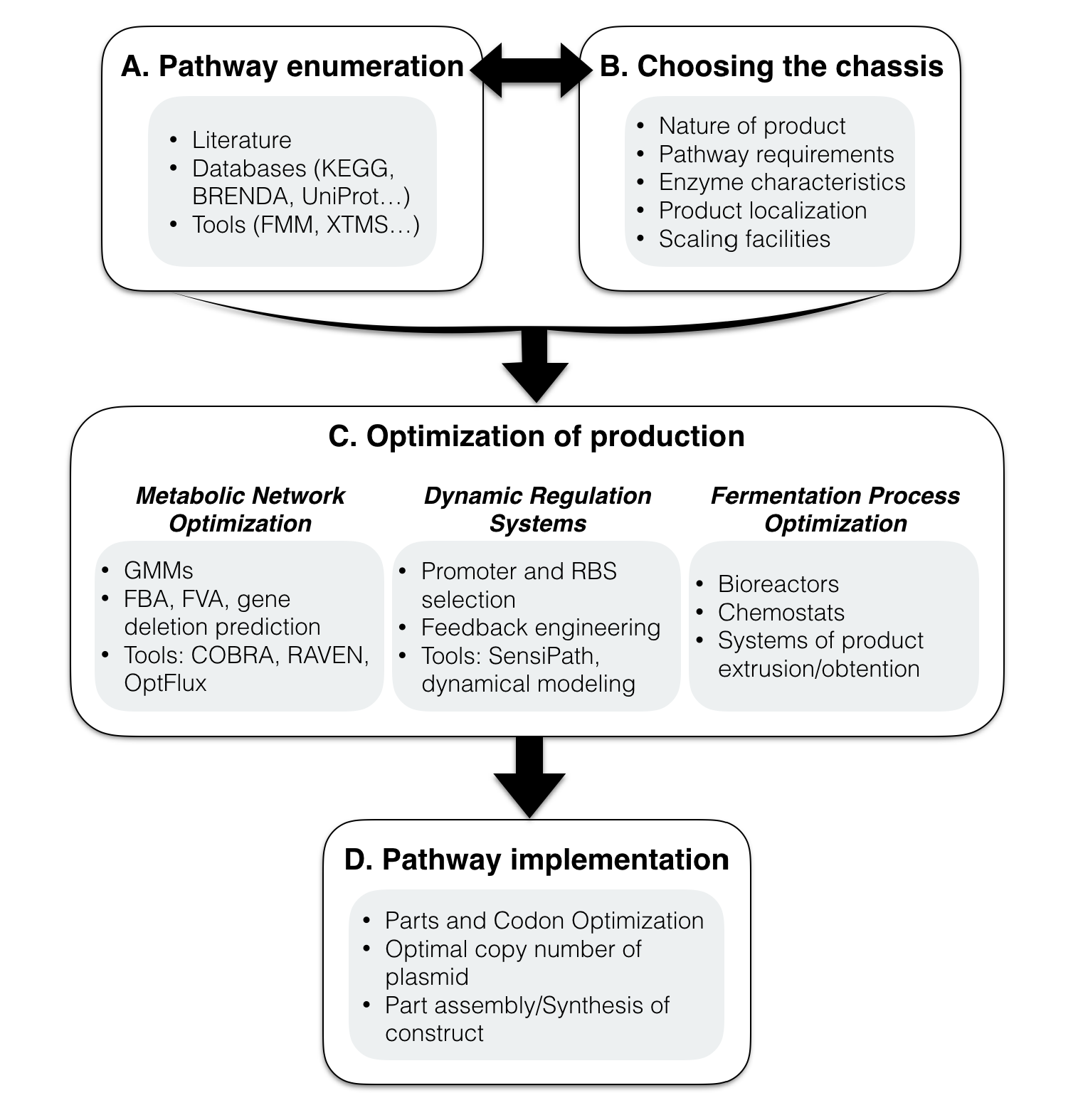
A. Pathway enumeration
First of all, depending on the purpose, the heterologous pathway producing the specific compound must be sought (Figure 1A). To do this, one might utilize personal knowledge of the metabolism along with literature and databases, in order to find the enzymes manufacturing the product of interest. However, there are some tools and databases which are dedicated to this task. In this direction, FMM (From Metabolite to Metabolite) [16] finds the possible pathway from KEGG database enzymes in order to produce a target metabolite from a given precursor. Moreover, XTMS (eXTended Metabolic Space) [17] enumerates pathways connecting the desired metabolite to the chassis metabolism, expands the scope of the possible pathway, scores them, and even discovers new reactions based on enzyme promiscuity for designing the pathways for unnatural compounds.
There are imperative points that have to be deliberated when designing the pathway: i) ensuring that the chassis is compatible with heterologous enzymes in cases such as enzyme post-translational modification (e.g. the glycosylation of the eukaryotic world which does not occur in prokaryotic cells) and codon usage, ii) verifying that well annotated mutant (when synthetic) and natural enzymes exist, iii) seeking the closest enzyme for a wanted substrate due to the large variability of substrate range for a given EC number in different organisms. For these purposes, there are informative databases easy to use. For instance, BRENDA [18] gives the different substrates associated with a given enzyme or EC number in different organisms, as well as inhibitors, kinetic values, mutant and recombinant version of the enzymes. KEGG [19] is useful to obtain different reactions and pathways associated with an enzyme. UniProt [20] contains the annotation of the genes from several organisms of a given enzyme and characteristic of the enzymes.
As a developing approach in synthetic biology, amending the enzyme and pathway efficiency or making the completely new enzyme activity using directed evolution or rational design, will increase the need for standard techniques to synthesize and screen the phenotypes. Furthermore, screening and selecting the best enzymes for an efficient pathway is being sophisticated by biosensor-based screening along with transcription factor and more recently RNA biosensors [21-24].
B. Choosing the chassis
Choosing the appropriate host for manufacturing a product, depends on i) the nature of the product and its required precursors, as well as their potential toxicity, ii) compatibility of the chassis cellular process to form the functional enzymes and pathway requirements, iii) whether the enzyme is prokaryotic or eukaryotic, iv) localization of the product, and v) scaling up the process in the future goals (Figure 1B).
Moreover, the host must have available genetic tools. For instance, Escherichia coli is the most known microorganism with well characterized cellular processes which could be used in order to develop new tools in synthetic biology, standardizing the methods as well as improving the production process. Other bacterial species, such as Bacillus subtilis and Pseudomonas putida, have attracted the interest of many scientists and engineers being more adaptable to industrial purposes and more suitable for several types of products. On the other hand, Saccharomyces cerevisiae has been welcomed as a promising cell factory carrying more developed cell processes, and more importantly, to perform simple eukaryotic post-translational modifications on enzymes and products. From an other aspect, S. cerevisiae could be easily assented for scaling up procedures. More recently, scientists have developed new chassis which have shown more adaptability especially for industrial conditions, including microorganisms able to utilize cheap substrates as a carbon source. Choosing the most compatible cell factory is a crucial step which has to be carefully investigated from the very beginning compeer with choosing the production pathway and its enzymes [25,26].
C. Optimization of bioproduction
Synthetic metabolic engineering does not only deal with implementing the heterologous enzymes in a chassis and obtaining the product. Optimizing cellular and environmental conditions is necessary to achieve an interesting production yield (Figure 1C). In this direction, three ways of optimization are described:
I) Metabolic network optimization
Genome-scale Metabolic Network Models (GMMs) representing stoichiometric whole cell metabolism are used to study, optimize and manipulate the cell metabolism [27]. To date, the GMMs of several organisms have been reconstructed and are freely available to download in SBML format [28]. To employ GMMs, one of the most used approaches is Flux Balance Analysis (FBA) [29]. Thereby, in metabolic engineering, GMMs can be applied for analyzing and manipulating the flux distribution in order to optimize the yield of the desired product. Obviously, for synthetic metabolic engineering, which goal is to produce a novel compound or overproduce a native compound through a synthetic pathway, first of all, metabolites and reactions associated with the pathway have to be added to the SBML file of the host’s metabolic network.
Once the GMM modified, the next step is to run multiple simulations in order to gain a profound insight on the pathway and its bottlenecks, and optimal growth condition while the product is manufactured [30,31]. To do this, as mentioned, FBA is the main tool which finds the balanced flux distribution in the metabolic network from the feeding sources downward to the objective function of the simulation. The objective function is a reaction of the metabolism set to be maximized. Biomass is a hypothetical reaction representing the growth rate of the cell in which all the precursors of the cellular dry weight are substrates of this reaction. Also, there are FBA derivatives such as FVA (Flux Variability Analysis) giving the conscious range of flux for all GMM reactions while the objective function is maximized. OptKnock and evolutionary algorithm are the other derivatives of the FBA finding the mutant by which the growth rate and product flux are optimized simultaneously [27].
Eventually, comparing several types of simulations on GMM provides a perspective on i) the best carbon source, ii) optimum growth condition e.g. oxygen level and iii) pathway bottlenecks to evaluate gene deletion and/or overexpression (see the case study and the wiki for the practical procedures). In order to perform these operations, there are popular, promising and easy to use available toolboxes such as COBRA toolbox [27], RAVEN toolbox [32] and OptFlux [33], with user manual to accomplish from very beginner levels to professional tasks.
II) Dynamic regulation systems
Natural biological feedback processes provide dynamic regulation and metabolic optimization, through controlling activation or inactivation of genes expression. In the past few years, synthetic circuits have been constituted to integrate metabolic and gene expression levels connecting and regulating the synthetic metabolic pathway tightly into the cell metabolism and more importantly to the cell gene expression and regulation network [34,35].
Metabolic network optimization solely remarks the systems as an enormous stoichiometric matrix. However, the enumerated synthetic pathway and intrinsic genetic modifications have to be solved into the host cellular processes. This negates the stress on cell equilibrium, and also maximizes the in vivo product yield. Moreover, this brings most of the theories achieved in the metabolic network optimization to practice.
For constructing such a system, first, the most effective precursor of the pathway has to be chosen. This precursor essentially must be the precursor of the pathway which locates at the bottleneck of the pathway to perform very effective dynamic regulation. Generally, these metabolites associate with specific transcription factors, used to trigger sensor responses. Then, the genes coding for enzymes catalyzing the precursor’s upstream reactions have to be constructed under promoters responsive to these transcription factors. Optionally, orthogonal repressors can be implemented to control the genes encoding the precursor’s downstream reaction enzymes [36]. Such repressors would be expressed under the control of the same biosensors, thus reversing the activating effect of the promoter for downstream enzymes (see Case Study). Therefore, this dynamic regulator increases the carbon flux to the final product not being toxic for the cell by expressing the enzyme at the certain required amount. Furthermore, such a system amends the product yield with Le Chatelier’s effect of chemical equilibrium [35].
As mentioned, the biosensor is built from the main precursor of the pathway [37,38]. Commonly, the main precursor locates at the branch of central metabolism toward the synthetic pathway. For these points at the cell metabolism, oftentimes, specific transcription factor could be found in some organisms. To seek this, a recent tool, SensiPath [39,40] has been made to wisely search for the transcription factor responding to a given metabolite. Moreover, assessing the strength of the promoters expressing enzymes and engineering them is a key point to reach the acceptable dynamic regulated system, thus higher yield.
When the parts and their positioning are defined, simulating the system can predict its behavior over time. Then, in the design-model-test cycle, the promoter strengths, RBSs and other variables can be tuned to get the optimal yield of the product, considering the usage of the cellular resources for enzyme production and growth. In order to model the genetic-metabolic circuits (dynamic regulation system), several kinds of methods could be applied. These methods should be linked to dynamical modeling, being stochastic/deterministic or continuous/discrete depending on the particular case and goal priorities. Similarly, the model could use paradigms such as ordinary or partial differential equations, network dynamics or agent interactions [41,42].
III) Fermentation process optimization
Optimization of the fermentation process is also a key step to achieve the maximum product yield. There are different operational modes to be used in bioreactors, such as batch, fed-batch or continuous, which determine the evolution of cell culture over time [43]. Depending on the goal, the bioprocess should be design with one bioreactor type or the other.
For instance, fed-batch cultures are very common, but using continuous-stirred tanks could be favorable for metabolic engineering purposes [44]. Since conditions reach a steady state and side-parameters do not vary over time, continuous systems are attainable to be characterised. They often use chemostats, which maintain constant volume on the tank and facilitate the assessment of metabolites [45]. Besides, bioprocesses of all types can be designed in a stepwise fashion, allowing control of precursors and intermediates concentration along the production pathway and the way of feeding the medium.
Accuracy in these combinations can provide ease of tuning towards the total optimization of the cell; whether maximising cell growth and precursor accumulation, or by the separation of the bioprocesses in different steps.
D. Implementing the pathway and its associated parts
Once the pathway has been enumerated, theoretically optimized and dynamically regulated, its genetic parts have to be built and cloned into the chosen chassis (Figure 1D). This procedure strongly depends on the chassis compatibility with the synthetic biology tools. Before that, the parts have to be adapted with host cellular machinery system and the genes codon optimized to be fully functional in the chassis. Then, the gene parts have to be designed, and synthesized or purchased. Toward assembling the defined genetic parts, two most used general approaches, Gibson assembly [46] and Golden Gate [47] and their similar and derived methods [48] could be used (or BioBrick Assembly especially in the iGEM competition) in well-known cell cloning factories such as E. coli. Rather than assembling the parts, the recently welcomed alternative way is to synthesize the whole constructs and transforming them into the cell directly [49]. Daily plunging in the price of gene synthesis is dramatically widening the usage of this admirable tool as an exceedingly faster-cheaper-better road.
Case study: PLA bioproduction
Poly-Lactic Acid (PLA) is a polymer of lactic acid with a wide range of applications due to its properties as biodegradable plastic. Frequently, its synthesis combines biological and chemical processes, the latter being expensive and detrimental for environment [50,51].
In 2010, Y.K. Jung, S. Y. Lee and their colleagues produced PLA by engineering E. coli [15]. They reported the heterologous biosynthesis of the PLA homopolymer and its copolymer, poly(3-hydroxybutyrate- co-lactate) or P(3HB-co-LA). However, when reviewing recent approaches at industrial scale, at the moment of the study only the enterprise Carbios [52] reported PLA manufactured solely biologically.
The assumption presented here is that metabolic optimization of PLA biosynthesis would foster its implementation on the bioplastic industry. Thus, during iGEM 2016 competition, the team Évry applied the methodology previously described for studying bioproduction of PLA. The following sections refer to the four presented steps, each including details and specifications concerning the PLA case.
A. Basic pathway: Pct and PhaC engineered enzymes
Following the article from Jung et al. [15], two genes were described as essential for PLA biosynthesis: an engineered Propionate CoA transferase (Pct) encoding for an enzyme which uses lactate as subtract and converts it into Lactyl-CoA, and an engineered PHA synthase (PhaC) which enzyme can polymerize monomers of Lactyl-CoA into PLA.
- Engineered Pct (Pct*): The wild type form of Pct, present in Clostridium propionicum, catalyzes the formation of propanoyl-CoA from propanoate. The introduction of the amino acid mutation A243T was found to efficiently convert lactate into lactyl-CoA.
- Engineered PhaC (PhaC*): Pseudomonas sp. MBEL 6-19 PHA synthase 1 is the original enzyme from which they performed four amino acid substitutions: E130D, S325T, S477F, and Q481K. The engineered version had enhanced activity towards (D)-lactyl-CoA and allowed its polymerization.
By having lactate as precursor, any bacterial chassis with these two functional enzymes would be expected to produce PLA. The following procedure was the analysis of candidate chassis that could provide a proper synthesis efficiency.
B. Pseudomonas putida, the best candidate chassis
As mentioned, synthetic bioproduction of PLA was already developed in E. coli [15]. However, we determined that several criteria could set other organisms as better chassis for such heterologous production.
First, considering the natural presence of precursor, species with high lactate yield were listed and highlighted:
- Wild type Lactobacillus casei RL20: Its production yield is 72 g/L at 48h, reaching 144.2 g/L at 48h when expressing the genes Pfk and Glk [53] .
- Bacillus subtilis MUR1: It can produce 99.3 g/L and 183.2 g/L of L-lactic acid in 12h and 52h respectively, with a maximum L-lactic acid production rate of 16.1 g/L/h [54]
- Pseudomonas putida: Good yield results of lactic acid have been observed from the activities of its iLDH (22.1 nmol/min*mg for L-isomer and 66.6 nmol/min*mg for D-isomer) [55]
Afterwards, several characteristics of the necessary enzymes were analyzed. On one hand, their original forms were both coming from prokaryotic bacteria [15]: this would assume bacterial chassis to be more suitable than others, i.e. yeast, which may use different machinery for post-translational modifications. On the other hand, the polymerization reaction could be a pathway bottleneck to overcome. Therefore, organisms that naturally produce polyesters similar to PLA, would provide better reaction efficiency. Pseudomonas spp., bacteria able to synthesize Polyhydroxyalkanoates (PHA)[56], could be an example fitting the two criteria.
However, as aiming to genetically modify the chassis, two additional criteria had to be considered: ease of manipulation and safety. For that, it should be a GRAS (Generally Recognized As Safe) bacterium with well described metabolism and commonly used for synthetic biology purposes. Finally, it was concluded that P. putida KT2440 would be the most suitable chassis for obtaining our PLA because of being a lactate producer efficient at polymerization [55,56], and being a GRAS strain widely used as work-horse for bioproduction [57].
C. Optimization of PLA bioproduction
Once conceived a basic design for PLA bioproduction in P. putida KT2440, three optimization approaches were used for improving the theoretical design: metabolic network optimization, dynamic regulation systems and fermentation process optimization.
I) Metabolic network optimization
A FBA was reported on P. putida KT2440, analyzing the flux distribution and thus improving the PLA yield [58]. A synthetic pathway with the reactions of exogenous Pct* and PhaC* and their corresponding metabolites was implemented in the GMM. The final PLA-producing P. putida KT2440 metabolic network contained 962 genes, 980 reactions and 899 metabolites.
During the optimization process, glucose and fructose were tested as substrates with two objective functions: PLA producing reaction and Biomass, the latter being a hypothetical reaction in which the flux is identical to cell growth rate. The implementation of PLA as a precursor of biomass was also studied to obtain a more realistic view on cell growth and PLA production simultaneously. All FBA experiments were performed using OptFlux toolbox [59].
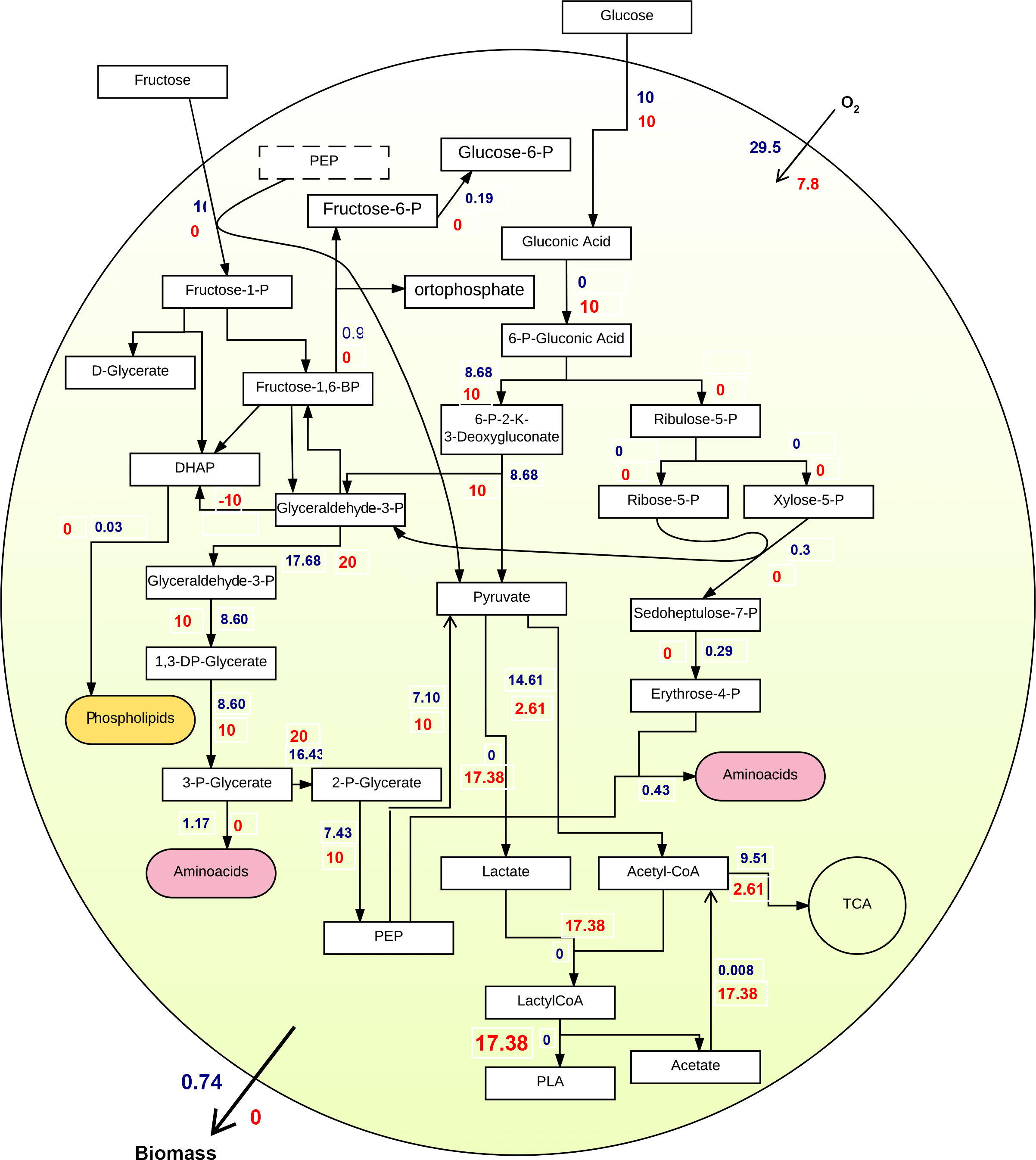
First experiment: Glucose was set as the substrate. Figure 2 shows two independent FBA on biomass (blue fluxes) and PLA producing reaction (red fluxes) as objective functions, using glucose as sole carbon source. Due to a biased optimization of FBA, the yield of PLA production equals to zero when the biomass is maximized, and vice versa. Besides, a comparison of flux distribution in the central metabolism of these two independent FBA demonstrates that the main bottleneck of PLA production locates in pyruvate fermentation to lactate.
Second experiment: It differed from the first experiment as fructose was the sole carbon source. As illustrated in Figure 3, flux distributions and flux values are different than pictured in Figure 2. More importantly, both biomass and PLA fluxes increased, compared to the previous experiment. Thus, FBA suggested that fructose is a more suited substrate to promote both growth and PLA production.
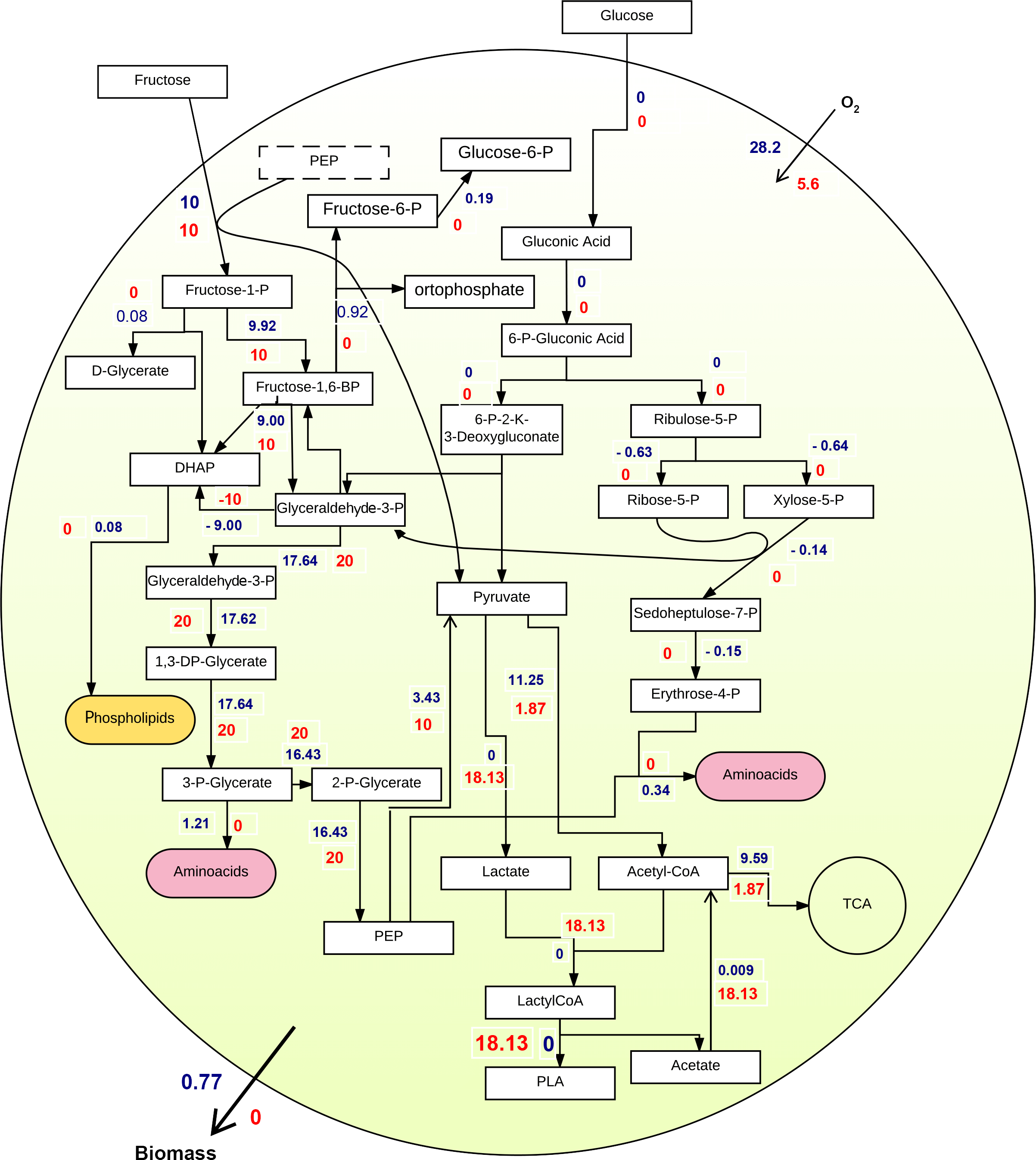
Third experiment: Finally, both Biomass and PLA were integrated at the same time into one objective function to have PLA as part of the cellular biomass. The stoichiometric ratio of PLA with regards to the whole biomass was put the same as other polymers of P. putida KT2440 GMM. The integration shown in Figure 4 enabled further investigation of the pathway and provided more realistic perspectives on the whole cell metabolism, emphasizing on PLA production while keeping an economic growth rate.

In growth conditions, the flux of the fermentation pathway was approximately zero. As a result, lactate supply was detected as being a main challenge in PLA production. In this direction, a useful mutant of lactate dehydrogenase (LDH) enzyme, d-LDH*, was found in the literature [60] enabling the use of both NADH and NADPH efficiently and giving access to a higher substrate consumption. The implementation of this mutant would be particularly necessary for PLA production optimization.
On the other hand, the oxygen uptake flux for PLA production, when used as the objective function, was 6-fold less consumed than when optimizing biomass in FBA. That indicated that low levels of oxygen would be sufficient for PLA production. Indeed, lower oxygen levels reduce biomass production and leads to carbon transformation into lactate via fermentation process and finally leads to PLA formation. However, due to the necessity of cellular biomass as the cell factory, the best solution would be to design a two step fermentation: first, the oxygen level would be set up with high aeration to increase the biomass; then, microaerobic conditions would be used to redirect the most of the carbon and energy into production of PLA.
Finally, in terms of carbon source, the in silico experiments indicated the use of fructose as carbon source should be prioritized over glucose for PLA production. Further experimental tests of growth rates should be performed in order to reassure the fructose employment significance shown by FBA.
A more detailed description of the experiments and analysis on the results can be found in the wiki FBA modeling page: http://2016.igem.org/Team:Evry/Model/FBA.
II) Dynamic regulation using biosensors
For optimizing PLA production, a feedback system depending on a lactate biosensor and repressible promoters was conceived, that would regulate the expression of our d-LDH*, Pct* and PhaC* genes. The designed system has synthetic regulation and improves the ratio PLA produced / enzyme needed. More precisely, the system increases the PLA yield by controlling the carbon flux of the pathway and the precursor toxicities in accordance with Le Chatelier’s principle, avoiding gene overexpression. As shown in Figure 5, it relies on two main mechanisms of regulation: an LldR system and a McbR system.

LldR system: The LldR responsive promoter has been well described in coli for regulation of lldPRD operon [61]. It works as a biosensor of lactate. In the PLA system, the LldR responsive promoter controls the expression of the operon with genes responsible for PLA production: PhaC* and Pct*. As a consequence, they are only transcribed when there is lactate in the cell. The gene encoding the LldR transcription factor is expressed under a constitutive promoter, provoking repression of the LldR responsive promoter in basal conditions.
McbR system: The McbR promoter is part of the TetR-family repressors, widely used in synthetic biology [62]. The repressible promoter is basally active, but it is inhibited when McbR TF is transcribed. In the PLA system, an McbR repressor is implemented as promoter of the LDH gene. Besides, a LldR responsive promoter regulates McbR gene expression: in the presence of lactate, it starts expressing the McbR protein, creating a feedback system.
This system was modelled to observe and predict the dynamics generated by different elements. Two strategies were used: agent-based modeling and differential equations modeling in the system represented as genetic circuit.
Kappa model: Agent-based modeling represents stochastic systems where agents and their interactions are defined [63]. In the model, the dynamics that the responsive elements would present if implemented on experimental lab were studied. The objective was to get to know the optimal combination of element variable features (ex. RBS strength) on the feedback loops to optimize PLA production. Because of having several elements to represent, interactions between the elements, and parameters that could be approximated using rate probabilities, Kappa language [64] – which uses agent-based modeling – was considered adequate for its implementation.
Dynamic modeling using differential equations: The interaction of the subsystems from a designed biosensor-based regulon can be translated into differential equations of the evolution of each component of the system. In the PLA system, these equations were designed based on mass action law, representing the different components of the genetic-metabolic circuit. Solving these equations demonstrated the evolution of each component time-proportionally.
Using a Kappa agent-based model, several simulations were run testing variations in agent reaction rates and LldR system was found to be the key factor. When tuning the promoter and RBS strength on LldR, so on its mRNA transcription and translation rates, different ratios PLA/Lactate were observed. In the optimal case, as shown in Figure 6B, was figured out setting a weak RBS strength.
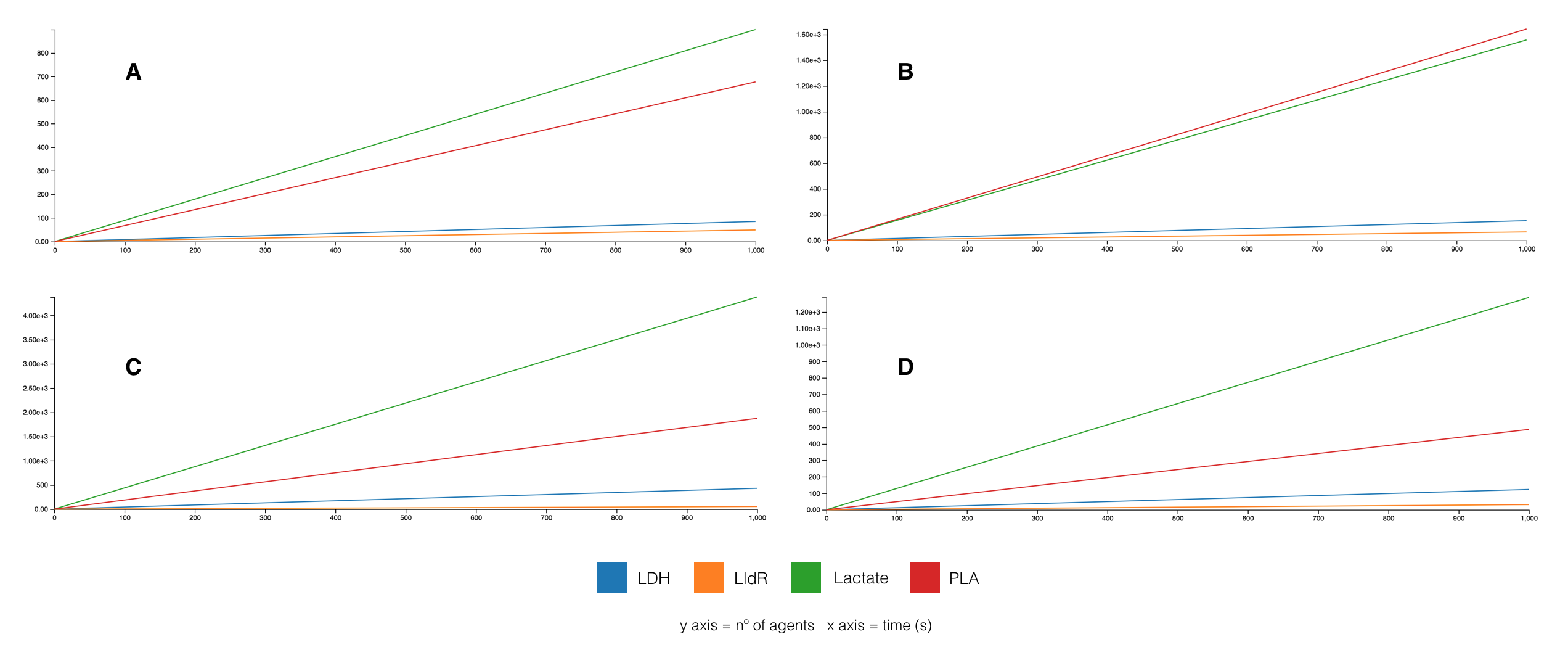
In the second model, differential equations of the dynamics of each component were extracted using a mathematical method from Brian Ingalls lab, University of Waterloo [65]. All the constants were set to 1, as the aim was to show how to extract equations related to the synthetic dynamic regulation system and observe its approximated behavior. They were solved using Python (equations and its code are described on the corresponding wiki section). Figure 7 shows that PLA production (red curve) increases while all the other components reach a balance after a period of time (in seconds), demonstrating that the evolution system works maximizing PLA production even using the constant parameter approximation.

The detailed construction description of both models can be found in the wiki of the project, at the Dynamic Regulation section: http://2016.igem.org/Team:Evry/Model/Dynamic. Similarly, details on parameters definition are described on the wiki.
III) DIY Continuous Bioprocess
In order to optimize the fermentation process, a Do-It-Yourself (DIY) bioprocess that would allow PLA production in a continuous fashion was conceived and constructed, as shown in Figure 8.

The whole bioprocess consisted on a DIY continuous pump, two bioreactors, one additional auxiliary reactor, a DIY-PLA-Extruder and a DIY-roller for final storage of the PLA product. Its main characteristic was the implementation of the continuous bioprocess in a stepwise manner which, by splitting the bioproduction in two chemostats, could induce progressively the crucial steps of the metabolic pathway.
Using chemostats
In this type of bioreactor the mass balance is described as indicated in eq.1. Once the steady state has been reached, the specific growth rate (µ) of microorganisms can be controlled. The steady flow (F) allows a system of a fixed volume (V), where accumulation or leakage of biomass in the system over time is null (dM/dt=0).

This characteristic allows cells to grow at a fixed specific growth rate (μ) for the achievement of a fixed value of biomass concentration (x) over time (t). This value can be controlled according to the Dilution time (D), which is equivalent to the Flow (F) per Volume (V) unit, that is D = F/V.

As a consequence, the amount of product can be maximized by increasing the cell concentration (x) and correlated to the flow optimization (F). According to this, the pumping system for the chemostats is optimized to provide a dilution time (D) never greater than the value of the maximum growth rate for P. putida under specific conditions (µmax in glucose: 0.212 h-1 = 0.0035min-1; µmax in glycerol: 0.206 h-1 = 0.0034min-1. Calculated from experiments.)
Optimal dilution times (Dopt < Dmax) can only be obtained by plotting the substrate consumption rate of the microorganism, which has not been assessed in this study.
Plotting growth rates and enabled dilution times under specific genetic modifications is a systematic approach for the step-wised maximisation of the production of specific products or metabolites (i.e. PLA or preceding precursors in preceding chemostats).
By using this approach, the effects of particular modifications can be assessed in specific steps in the metabolic pathway, since an increase in the productivity of a specific metabolite can be modeled and estimated in vivo in a single chemostat. The more chemostats, the more precursors to be studied throughout the bioprocess; providing a better optimisation of the metabolic pathway.
The bioprocess was manufactured in a DIY manner, to show its implementation would be affordable. Moreover, there were possible improvements in the mechanical system of extrusion and storage: a PLA extruder, with the help of a heater, would allow ejection of raw PLA filaments which, at their turn, would roll over a roller, solidify and be stored.
Further details on the manufacturing of the system, mathematical modeling of the bioprocess and steady flow testing are described on the wiki section of bioprocess: http://2016.igem.org/Team:Evry/Improvements/Bioreactor
D. Implementation
To achieve a correct implementation and expression of our genes, works of well-known laboratories using Pseudomonas spp. as engineering chassis were studied. Advised by Victor de Lorenzo (CNB-CSIC, Madrid, Spain), the best option was set to be using Standard European Vector Architecture plasmids (SEVA). The plasmid construction was set as follows:
For the implementation of the basic operon, the choice would be an inducible promoter by IPTG, as it is well known and regulable. In terms of antibiotic, P. putida is naturally resistant to Chloramphenicol, so an alternative resistance gene such as Kanamycin would be used.
Next step would be the insertion of LDH encoding gene, to foster lactate production as described on optimization. The best strategy would consist in using another inducer to regulate the lactate production the and induction of PLA genes. Cyclohexanone (CH) would be a good example of inducer, combined with Spectinomycin resistance.
Besides, the selection of an optimal RBS for P. putida should be taken into account. If implementing the dynamic regulation system, the two plasmid systems described would be modified by adding extra elements of control, as shown in Figure 5.
Finally, it would be necessary to include in our gene design the overhangs or necessary bases needed for the assembly method chosen. For the PLA production case, the choice was the Standard BioBrick Assembly, so its characteristic Prefix and Suffix would be required, as well as checking absence of the restriction sites (EcoRI, PstI, SpeI, XbaI) in the gene sequence.
Conclusion
In this report, we presented a well-organized plan for synthetic metabolic engineering. Following this protocol enables one to design an elaborate experiment through a standardized protocol for future research and industrial purposes. Our approach brings together two distinct disciplines related to cell engineering: synthetic biology and systems biology. This integration has been neglected despite massive progress in synthetic biology and systems biology separately [66, 67]. This protocol was followed by our iGEM team with a case study on PLA production to build a platform for future studies in this era of bioproduction.
Since some parts of the methodology had to go more in details, continuing each step with a PLA example guides to the procedure has to be done for any arbitrary project. Even though this project did not succeed in the wet lab experiment, the main goal was achieved in the integration of several tools to present a cohesive protocol validated by judging comments on that. Participation of several students from different backgrounds facilitated the iGEM team to get to this destination.
The perspective of this report is to accomplish more combination in the daily-used tools of biotechnology, systems and synthetic biology. This will negate obstacles in bioproduction such as i) expensive inducers for biochemical production, ii) lack of the enzymes and pathways for manufacturing the unnatural products iii) improving the yield through several optimization processes [68,69].
Response to Reviewers
A transcript of the reviewer comments and author responses from the Live Peer Review Jamboree can be found here: Evry Response to Reviewers
References
- Leduc S. La biologie synthétique. A. Poinat, Paris; 1912.
- Loeb J. The mechanistic conception of life. Biological essays. University of Chicago Press, Chicago; 1912.
- Cameron DE, Bashor CJ, Collins JJ. A brief history of synthetic biology. Nat Rev Microbio. 2014: 1-10. doi: 10.1038/nrmicro3239
- Peretó J. Erasing Borders: A Brief Chronicle of Early Synthetic Biology. J Mol Evol. 2016. doi: 10.1007/s00239-016-9774-4
- Wittmann C, Lee SY, editors. Systems metabolic engineering. Springer Science & Business Media; 2012 Jun 15.
- Lee JW, Na D, Park JM, Lee J, Choi S, Lee SY. Systems metabolic engineering of microorganisms for natural and non-natural chemicals. Nat Chem Bio. 2012 Jun 1;8(6):536-46.
- Purnick PE, Weiss R. The second wave of synthetic biology: from modules to systems. Nat Rev Mol Cell Bio. 2009 Jun 1;10(6):410-22.
- Keasling JD. Synthetic biology and the development of tools for metabolic engineering. Metab Eng. 2012 May 31;14(3):189-95.
- Nielsen J, Keasling JD. Synergies between synthetic biology and metabolic engineering. Nat Biotech. 2011 Aug 1;29(8):693-5.
- McAdams HH, Arkin A. Towards a circuit engineering discipline. Curr Biol. 2010;10: R318–R320.
- McMillen HD, Collins JJ. Engineered gene circuits. Nature. 2002; 420: 224–230. doi: 10.1038/nature01257; pmid: 12432407.
- Maarleveld TR, Khandelwal RA, Olivier BG, Teusink B, Bruggeman FJ. Basic concepts and principles of stoichiometric modeling of metabolic networks. Biotechnol J. 2013; 8: 997–1008.
- Ang J, Bagh S, Ingalls BP, McMillen DR. Considerations for using integral feedback control to construct a perfectly adapting synthetic gene network. J Theor Bio. 2010;266:723–738.
- Lechner A, Brunk E, Keasling JD. The Need for Integrated Approaches in Metabolic Engineering. Cold Spring Harb Perspect Biol 2016;8:a023903.
- Jung YK, Kim TY, Park SJ, Lee SY. Metabolic Engineering of Escherichia coli for the Production of PLA and Copolymers in E. coli. Biotech and Bioeng. 2010; 105(1): 161-171.
- Chou CH, Chang WC, Chiu CM, Huang CC, Huang HD. FMM: a web server for metabolic pathway reconstruction and comparative analysis. Nucleic Acids Res. 2009 Jul 1;37(suppl 2):W129-34.
- Carbonell P, Parutto P, Herisson J, Pandit SB, Faulon JL. XTMS: pathway design in an eXTended metabolic space. Nucleic Acids Res. 2014 Jul 1;42(W1):W389-94.
- Chang A, Schomburg I, Placzek S, Jeske L, Ulbrich M, Xiao M, Sensen CW, Schomburg D. BRENDA in 2015: exciting developments in its 25th year of existence. Nucleic Acids Res. 2014 Nov 5:gku1068.
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016 Nov 28:gkw1092.
- UniProt Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2014 Oct 27:gku989.
- Rogers JK, Church GM. Genetically encoded sensors enable real-time observation of metabolite production. Proc Natl Acad Sci. 2016 Mar 1;113(9):2388-93.
- Eggeling L, Bott M, Marienhagen J. Novel screening methods—biosensors. Curr Opin Biotech. 2015 Dec 31;35:30-6.
- Mustafi N, Grünberger A, Mahr R, Helfrich S, Nöh K, Blombach B, Kohlheyer D, Frunzke J. Application of a genetically encoded biosensor for live cell imaging of L-valine production in pyruvate dehydrogenase complex-deficient Corynebacterium glutamicum strains. PLoS One. 2014 Jan 17;9(1):e85731.
- Schallmey M, Frunzke J, Eggeling L, Marienhagen J. Looking for the pick of the bunch: high-throughput screening of producing microorganisms with biosensors. Curr Opin Biotech. 2014 Apr 30;26:148-54.
- Lee SK, Chou H, Ham TS, Lee TS, Keasling JD. Metabolic engineering of microorganisms for biofuels production: from bugs to synthetic biology to fuels. Curr Opin Biotech. 2008 Dec 31;19(6):556-63.
- Yadav VG, De Mey M, Lim CG, Ajikumar PK, Stephanopoulos G. The future of metabolic engineering and synthetic biology: towards a systematic practice. Metab eng. 2012 May 31;14(3):233-41.
- Schellenberger J, Que R, Fleming RM, Thiele I, Orth JD, Feist AM, Zielinski DC, Bordbar A, Lewis NE, Rahmanian S, Kang J. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2. 0. Nat Protoc. 2011 Sep 1;6(9):1290-307.
- Feist AM, Herrgard MJ, Thiele I, Reed JL, Palsson BØ. Reconstruction of Biochemical Networks in Microbial Organisms, Nat Rev Microbiol. 2009 Feb;7(2):129-143.
- Orth JD, Thiele I, Palsson BØ. What is flux balance analysis?. Nat biotechnology. 2010 Mar 1;28(3):245-8.
- Fehér T, Planson AG, Carbonell P, Fernández‐Castané A, Grigoras I, Dariy E, Perret A, Faulon JL. Validation of RetroPath, a computer‐aided design tool for metabolic pathway engineering. Biotechnology journal. 2014 Nov 1;9(11):1446-57.
- Fernández-Castané A, Fehér T, Carbonell P, Pauthenier C, Faulon JL. Computer-aided design for metabolic engineering. Journal of biotechnology. 2014 Dec 20;192:302-13.
- Agren R, Liu L, Shoaie S, Vongsangnak W, Nookaew I, Nielsen J. The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput Biol. 2013 Mar 21;9(3):e1002980.
- Rocha I, Maia P, Evangelista P, Vilaça P, Soares S, Pinto JP, Nielsen J, Patil KR, Ferreira EC, Rocha M. OptFlux: an open-source software platform for in silico metabolic engineering. BMC Systems Bio. 2010 Apr 19;4(1):1.
- Venayak N, Anesiadis N, Cluett WR, Mahadevan R. Engineering metabolism through dynamic control. Curr Opin Biotech. 2015 Aug 31;34:142-52.
- Xu P, Li L, Zhang F, Stephanopoulos G, Koffas M. Improving fatty acids production by engineering dynamic pathway regulation and metabolic control. Proc Natl Acad Sci. 2014 Aug 5;111(31):11299-304.
- Stanton BC, Nielsen AA, Tamsir A, Clancy K, Peterson T, Voigt CA. Genomic mining of prokaryotic repressors for orthogonal logic gates. Nat Chem Bio. 2014 Feb 1;10(2):99-105.
- De Paepe B, Peters G, Coussement P, Maertens J, De Mey M. Tailor-made transcriptional biosensors for optimizing microbial cell factories. J Ind Microbiol Biotechnol. 2016 Nov 11:1-23.
- Morgan SA, Nadler DC, Yokoo R, Savage DF. Biofuel metabolic engineering with biosensors. Curr Opin Chem Bio. 2016 Dec 31;35:150-8.
- Libis V, Delépine B, Faulon JL. Expanding biosensing abilities through computer-aided design of metabolic pathways. ACS Synth Bio. 2016 Mar 30.
- Delépine B, Libis V, Carbonell P, Faulon JL. SensiPath: computer-aided design of sensing-enabling metabolic pathways. Nucleic Acids Res. 2016 Apr 22:gkw305.
- Machado D, Costa RS, Rocha M, Ferreira EC, Tidor B, Rocha I. Modeling formalisms in Systems Biology. AMB Express 2011, 1:45.
- Gorochowski TE. Agent-based modelling in synthetic biology. Essays Biochem. 2016;60: 325–336. DOI: 10.1042/EBC20160037
- Doran PM. Bio-process Engineering Principles. Elsevier, 2nd ed. 2013 ISBN 978-0-12-220851-5.
- Lin YH, Bayrock D, Ingledew WM. Metabolic flux variation of Saccharomyces cerevisiae cultivated in a multistage continuous stirred tank reactor fermentation environment. Biotechnol Prog. 2001 Nov-Dec;17(6):1055-60.
- Zhang Z, Boccazzi P, Choi HG, Perozziello G, Sinskey AJ, Jensen KF. Microchemostat-microbial continuous culture in a polymer-based, instrumented microbioreactor. Lab Chip. 2006;6(7): 906–13.
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009 May 1;6(5):343-5.
- Werner S, Engler C, Weber E, Gruetzner R, Marillonnet S. Fast track assembly of multigene constructs using Golden Gate cloning and the MoClo system. Bioengineered. 2012 Jan 1;3(1):38-43.
- Moore SJ, Lai HE, Kelwick RJ, Chee SM, Bell DJ, Polizzi KM, Freemont PS. EcoFlex: A Multifunctional MoClo Kit for E. coli Synthetic Biology. ACS Synth Bio. 2016 Apr.
- Kosuri S, Church GM. Large-scale de novo DNA synthesis: technologies and applications. Nat Methods. 2014 May 1;11(5):499-507.
- Sin LT, Rahmat AR, Rahman WA. Overview of Poly(lactic Acid). Polylactic Acid. Elsevier, 2012; 1-70. ISBN: 978-1-4377-4459-0
- Yang JE, Choi SY, Shin JH, Park SJ, Lee SY. Microbial production of lactate- containing polyesters. Microb Biotech. 2013; 6(6); 621–636.
- Carbios, Reinvent Polymers Lifecycle. Available from: http://www.carbios.fr/
- Gong Y, Li T, Li S, Jiang Z, Yang Y, Huang J, Liu Z, Sun H. Achieving High Yield of Lactic Acid for Antimicrobial Characterization in Cephalosporin- Resistant Lactobacillus by the Co-Expression of the Phosphofructokinase and Glucokinase. J Microbiol Biotechnol. 2016; 26(6):1148-61.
- Gao T, Wong Y, Ng C, Ho K. L-lactic acid production by Bacillus subtilis MUR1. Bioresour Technol. 2012; 121:105-110.
- Wang Y, Min L, Zhang Y, et al. Reconstruction of lactate utilization system in Pseudomonas putida KT2440: a novel biocatalyst forL-2-hydroxy- carboxylate production. Sci Rep. 2014; 4: 6939.
- Rehm BHA. Bacterial polymers: biosynthesis, modifications and applications. Nat Microbio. 2010; 8, 578-592.
- Nikel PI, Martinez-Garcia E, de Lorenzo V. Biotechnological domestication of pseudomonads using synthetic biology. Nat Rev Microbiol. 2014;12, 368-379.
- Nogales J, Palsson BØ, Thiele I. A genome-scale metabolic reconstruction of Pseudomonas putida KT2440: iJN746 as a cell factory. BMC Syst Biol. 2008;2:79.
- Rocha I, Maia P, Evangelista P, Vilaça P, Soares S, Pinto JP et al. OptFlux: an open-source software platform for in silico metabolic engineering. BMC Systems Biology 2010, 4:45.
- Meng H, Liu P, Sun H, Cai Z, Zhou J, Lin J, Li Y. Engineeringa d-lactate dehydrogenase that can super-efficiently utilize NADPH and NADH as cofactors. Sci Rep. 2016;6.
- Aguilera L, Campos E, Gimenez R, Badia J, Aguilar J, Baldoma L. Dual Role of LldR in Regulation of the lldPRD Operon, Involved inL-Lactate Metabolism in Escherichia coli. J Bact. 2008; 190(8): 2997–3005.
- Stanton BC, Nielsen AA, Tamsir A, Clancy K, Peterson T, Voigt, CA. Genomic mining of prokaryotic repressors for orthogonal logic gates. Nat Chem Bio. 2014; 10: 99-105.
- An G, Mi Q, Dutta‐Moscato J, Vodovotz Y. Agent‐based models in translational systems biology. Wiley Interdisciplinary Reviews: Systems Bio Med. 2009 Sep 1;1(2):159-171.
- Boutillier P, Feret J, Krivine J, Quyên LK. KaSim & KaSa reference manual (release 3.90). Available from: http://dev.executableknowledge.org/docs/KaSim-manual-master/KaSim_manual.htm
- Ingalls BP, Yi TM, Iglesias PA. Using control theory to study biology. Systems Model Cell Bio. 2006:243-267.
- King ZA, Lloyd CJ, Feist AM, Palsson BO. Next-generation genome-scale models for metabolic engineering. Current opinion in biotechnology. 2015 Dec 31;35:23-9.
- Stephanopoulos G. Challenges in engineering microbes for biofuels production. Science. 2007 Feb 9;315(5813):801-4.
- Yadav VG, De Mey M, Lim CG, Ajikumar PK, Stephanopoulos G. The future of metabolic engineering and synthetic biology: towards a systematic practice. Metabolic engineering. 2012 May 31;14(3):233-41.
- Abbott DA, Zelle RM, Pronk JT, Van Maris AJ. Metabolic engineering of Saccharomyces cerevisiae for production of carboxylic acids: current status and challenges. FEMS yeast research. 2009 Dec 1;9(8):1123-36.