iGEM REPORT: From Carbon Dioxide to Biofuel
Note: This iGEM Report was submitted to the PLOS iGEM Realtime Peer Review Jamboree, and has not undergone formal peer review by any of the PLOS journals. We welcome your comments on this work.
From Carbon Dioxide to Biofuel
Tore Bleckwehl (1), Sebastian Blunk (1), Sandra Brosda (1), Julian Droste (1), Annika Fust (1), Birte Hollmann (1), Boas Pucker* (1), Simon Riedl (1), Janina Tiemann (1), David Wollborn (1), Nils-Christian Lübke (1), Manuel Wittchen (1), Timo Wolf (1)
- Center for Biotechnology, Bielefeld University, Bielefeld, Germany
*Corresponding author: [email protected]
Author Contributions
Conceptualization: TB, SeB, SaB, JD, AF, BH, BP, SR, JT, DW
Investigation: TB, SeB, SaB, JD, AF, BH, BP, SR, JT, DW
Writing – Original Draft: SeB, SaB, JD, BP
Writing – Review & Editing: TB, SeB, SaB, JD, BH, BP
Visualization: SaB, AF
Funding Acquisition: AF, JT, DW
Supervision: NL, MW, TW
Abstract
Ecological power management is currently facing three major challenges: the storage of electrical power, the development of renewable energy resources and the increase of atmospheric carbon dioxide due to the use of fossil fuels. Especially, when considering the decreasing fossil fuel reserves and the increasing consumption, new solutions are urgently needed. Therefore, we developed a concept to face these three challenges by engineering Escherichia coli.
The concept comprises a biofuel production from carbon dioxide where the energy is supplied from external electricity. For the uptake of electrical power we developed an electrophilic E. coli that can be supplied with electrons by a mediator-based uptake and a feed-in loop within the citric acid cycle. The feasibility was tested in a self-made bioreactor.
Furthermore, the idea to complete the whole Calvin cycle in E. coli by characterizing the three missing enzymes, including the RuBisCO was tested. Additionally, we deployed a bacterial microcompartment from cyanobacteria for carbon dioxide fixation under aerobic conditions and characterized a Bio-bricked pathway of the isobutanol production to complete our concept.
Moreover, an antibiotic-free selection system based on the iGEM standard plasmid pSB1C3 was developed. This system could reduce the use of antibiotics within the iGEM community and beyond.
Financial Disclosure
Funding was provided by Merck KGaA, Stockmeier Holding GmbH, Promega GmbH, Evonik Industries AG, Baxter Oncology GmbH, Satorius Stedim Biotech GmbH, WeissBioTech GmbH, IIT GmbH (business unit BIEKUBA), C3 Prozess- und Analysentechnik GmbH, Fisher Scientific GmbH, PlasmidFactory GmbH & Co. KG, Center for Biotechnology (CeBiTec), Bielefeld University, Faculty of Technology at Bielefeld University, Faculty of Biology at Bielefeld University and SYNENERGENE. However, the funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
The authors have declared that no competing interests exist.
Data Availability
All data are fully available without restriction: http://2014.igem.org/Team:Bielefeld-CeBiTec.
Introduction
The major challenges of an ecological power management include the storage of electrical power, the development of renewable energy resources that are not timely and locally synced with demand, and the increase of atmospheric carbon dioxide. Furthermore, alternatives to fossil fuel are required as the amount of available crude oil is decreasing [1].
One of the biggest current challenges of an ecological power management is the transport and storage of electricity [2]. The use of regenerative sources like solar or wind power has risen significantly over recent decades. The problem of these sources is the fluctuating availability that leads to the fact that most of the required energy is still covered through traditional energy sources like coal or nuclear power. This means that the energy produced by renewable sources mainly acts as a surplus that cannot be stored efficiently at present. A further problem is that the lowest portion of the generated energy is needed at the production location and part of the energy will be lost during the transport to its destination. One idea to increase the efficiency of storage and transportation is the use of microorganisms. They can use the energy to generate biomass or other transportable and storable products. E. coli was selected in this project due to its superior accessibility by genetic engineering methods.
There are several approaches to supply microorganisms with electrons in order to support microbial respiration. One promising technique is the direct transfer of electrons to microorganisms. Electrotrophic bacteria are able to directly accept electrons from electrodes for the reduction of terminal electron acceptors. They are also called electrode oxidizing bacteria. The possibility of electron transfer to microorganisms was investigated for different species like Geobacter sp. [3]. The combination of biocatalysts and electrochemical reaction steps is potentially of great relevancy as bio-electrochemical processes can be supplied with redox-equivalents without the need of a co-substrate. Furthermore, electrons can be regarded as one of the cheapest redox equivalents available [4].
Direct electron transfer in bacteria is very complex and not completely understood so far. Therefore, indirect electron transfer based on a mediator that is reduced at the electrode within an electro biochemical reactor and reoxidized by bacterial cells is much more feasible [5]. E. coli as a Gram-negative bacterium has two membranes with a periplasmatic space between them, which has to be overcome for a successful electron transfer. Most promising mediators to address this challenge are neutral red, bromphenol blue and cytochromes. To supply the cells with electrons, different bioreactor concepts are available. One is to operate a separated batch reactor termed H-cell [4].
Another major energy-related problem is the increased quantity of carbon dioxide (CO2) in our atmosphere, which evolved, over the last centuries by the use of fossil fuels to generate energy. The annual global emission has increased by 80% between the years 1970 and 2004 [6] and climate change is considered nowadays as one of the biggest challenges for decades to come [7]. About 80% of the industrial emission results from combustion of coal, oil and natural gas. Several approaches to tackle this problem were already formulated to capture emitted CO2 in metals [8]. An additional factor leading to rising amounts of CO2 in our atmosphere is industrial livestock farming that generates methane and carbon dioxide as site products [9]. Moreover, the typical balance between consumption and production of CO2 is destabilized by a decreasing total forest area and increasing emissions [10]. Plants consume CO2 during the photosynthesis that is split into a light-dependent and a light-independent reaction, the Calvin cycle, also known as the reductive pentose pathway. The products of the light-dependent reaction, ATP and NADPH, are used by the Calvin cycle to produce higher sugars by simultaneous incorporation of CO2. However, some enzymes involved in this process are not limited to plants, but also present in different bacterial species.
Different approaches for an oxygen insensitive carbon dioxide fixation in E. coli have been investigated, like the 3-hydroxypropionate cycle [11], the use of a CO2 fixing microcompartment from cyanobacteria [12] and the carbon dioxide fixation with RuBisCO itself [13]. However, these approaches were carried out using different glycolytic carbon substrates as main source. By completing the Calvin cycle in E. coli with the three missing enzymes sedoheptulose-1,7-bisphosphatase (SBPase), phosphoribulokinase (PrkA), and ribulose-1,5-bisphosphate carboxylase/oxygenase (RuBisCO) we want to enable the cells to use inorganic carbon dioxide as the main carbon source. To avoid undesired side reactions of the RuBisCO in the presence of oxygen [14] and to achieve a more efficient CO2 fixation we wanted to introduce a bacterial microcompartment called carboxysome from Halothiobacillus neapolitanus [12]. Within this microcompartment, a CO2 enrichment is achieved in the presence of the carboxy anhydrase [12].
The CO2 fixation via the Calvin cycle generates 3-phosphoglycerate (PGA) [15]. This is transformed to pyruvate during the glycolysis. Pyruvate is an important substance in the central metabolism. It serves as a precursor for the synthesis of a variety of industrially relevant products like isobutanol, isoprene, putrescine, or even antibiotics. We selected isobutanol as the final product. The corresponding biosynthesis pathway starts with pyruvate and involves five enzymes of which three already exist in E. coli [16]. Applications of isobutanol include lube oil additives, conversion to isobutyl acetate, direct solvent, and conversion to amino resins, isobutylamines, or acrylate and methacrylate esters [16–19]. It can also be considered as a biofuel or an additive for petrol and is therefore a well suited product for this proof of concept study.
A general important aspect of synthetic biology is to prevent the uncontrolled interaction between the genetically modified organisms, the environment and mankind. Therefore, many projects lead to the development of sophisticated biosafety systems [20]. As antibiotic resistances are used in laboratories to select for successfully genetically modified organisms [21–24] our approach aims to reduce the spread of antibiotic resistances by controlling the bacterial cell division. Therefore, we implemented an antibiotic-free selection system by constructing BioBricks for the complementation of a D-alanine auxotrophic E. coli strain. In E. coli D-alaine is synthesized by two alanine racemases (Alr and Dadx). The absence of both alanine racemases leads to a strict dependence on D-alanine due to a missing crosslinkage of the peptidoglycan layer, which is normally performed between meso-diaminopimelic acid and D-alanine [25]. Since a lack of D-alaine has bacteriolytic effects we used its complementation by the alanine racemase for an antibiotic-free selection system and for molecular cloning without antibiotics.
In summary, we engineered an electrophilic E. coli strain together with an electro biochemical bioreactor. In addition, we constructed BioBricks for the carbon dioxide fixation in aerobic bacteria and for the isobutanol production in E. coli. Furthermore, we developed an antibiotic-free selection system based on the iGEM standard plasmid pSB1C3. Besides our new findings, we were able to reproduce and confirm several published results.
Materials and Methods
Basic molecular biology techniques
All detailed protocols used in this work are available online [26]. Therefore, we provide only brief descriptions of the important methods here. Descriptions of basic molecular biology techniques like cloning can be found exclusively online [26]. If not stated otherwise we used E. coli KRX (Promega) for the experiments.
Anaerobic cultivation
The anaerobic cultivation was carried out in gas-tight 15 mL tubes additionally sealed with parafilm. All media and buffer were degassed with nitrogen gas (N2) via a sparger before starting cultivation. Inoculation and sampling took place in a two-hand Glove Bag (Atmos Bag, Sigma Aldrich). The preculture was grown under aerobic condition at 37°C until it reached an OD600 of 0.6 to 0.8. Cells were washed two times with oxygen-free PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) and then again with degassed medium. The tubes were cultivated in a shaker at 37°C and 180 rpm. Sampling took place once a day. Growth was analyzed by measurement of OD600 and HPLC analysis of the supernatant was performed to measure fumarate, succinate and glucose. After sampling the tubes were overflowed with nitrogen gas, tightly closed and sealed with parafilm again for further cultivation at 37°C.
Biolog analysis
The strains to be analyzed were grown at 37°C overnight on a BUG+B (Biolog Universal Growth Medium + 5% sheep blood) agar plate by streaking out for single colonies. Single colonies were picked with a sterile swab and transferred into a sterile tube containing 10 mL of 1x IF-0a solution to adjust transmittance of 42%. 1.8 mL of this suspension was mixed with 9 mL solution B (144 µL dye mix A for Gram-negative bacteria, 1.856 mL ddH2O, 8 mL 1.2x IF-oa solution). Afterwards, 100 µL of this mixture were loaded in each well of the Biolog PM plate (96 wells). The plate was incubated for 48 h in the OmniLog PM system (Biolog, Inc.).
H-cell reactor cultivation
The electrochemical behavior of engineered E. coli strains was analyzed in a separated H‑shaped batch reactor. In this set up two bottles are connected by a flange with an ion exchange membrane in the middle, separating the anodic and cathodic chambers. As the separator a polymer electrolyte Nafion proton exchange membrane was used. A detailed pretreatment protocol is available online [26]. The reactor was operated with a three-electrode system. This system contains a working-, a counter- and a reference electrode which allows measurement and setting of a specific potential at the working electrode. A silver/silver chloride electrode served as reference with a known constant potential versus a standard hydrogen electrode. For the control of the voltage difference between the working and the reference electrode a potentiostat from the company Gamry® was used. This device allowed to perform cyclic voltammetry assays for the characterization of mediated electron transfer.
The strains to be analyzed were grown at 37°C overnight in minimal M9 medium with 50 mM xylose as carbon source. The cathodic space of the H-cell reactor was filled with 500 mL M9 medium (50 mM xylose) supplemented with neutral red to a final concentration of 100 µM. Previous to inoculation the medium was degassed with nitrogen gas. The anodic space was filled with phosphate buffer. The cultivation started by inoculation to an OD600 of 0.1 from the pre-culture at 37°C and an airflow of 0.75 standard liters N2 per minute. The experiments were performed with scan limits from -800 mV to 700 mV at different scan rates and step limits. Additionally, different electrode materials (carbon fleece, carbon fabric and platinum) and sizes were tested.
Seduheptulose-1,7-bisphosphate assay
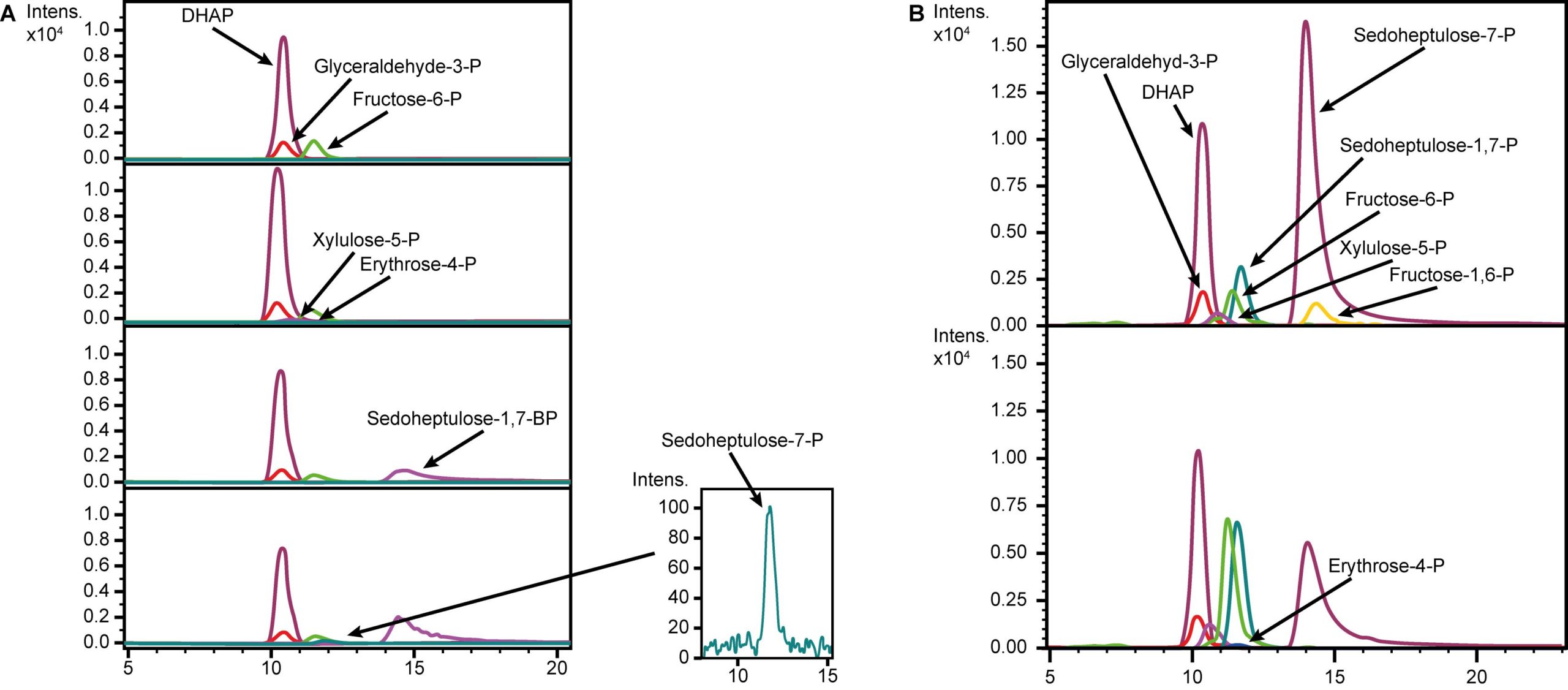
The validation of the seduheptulose-1,7-bisphosphatase (SBPase) activity was carried out based on the description of the initial enzyme characterization [27]. Required assay substrates were not commercially available. Therefore, transketolase (Tkt) and fructose bisphosphate aldolase (Fba) were used for the substrate formation. The enzymes Tkt, Fba and SBPase were expressed in E. coli KRX using pET16 expression plasmids with 100 µM IPTG for induction. Enzymes were purified via His-tag. The enzyme concentrations were adjusted based on Bradford assay results. The negative control was performed by adding no enzyme to the substrates to ensure that all observed products in the other reactions were generated by Tkt, Fba and SBPase. The assay itself consisted of three reactions. The first reaction included the Tkt that catalyzed the reaction of fructose-6-posphate (F6P) and glyceraldehydes-3-phosphate (GAP) to erythrose-4‑phosphate (E4-P). For the second reaction Fba was added to convert E4-P with DHAP to sedoheptulose-1,7-bisphosphate (SBP). In the last reaction the SBPase (GlpX) was added resulting in the final product, sedoheptulose-7-phosphate (S7-P).
RuBisCO assay
The assay was carried out based on published descriptions [28]. Crude protein extracts were incubated at 37°C in presence of ribulose-1,5-bisphosphate (Ru-BP) and CO2 was gassed in constantly. Samples were taken every five minutes and analyzed for changes in substrate and product concentrations via HPLC.
SDS-PAGE and MALDI-TOF-MS
Cultures of E. coli with the designated plasmid were cultivated at 37°C. Gene expression of the plasmid was induced at OD600 0.6 to 0.8 by adding 1 mM IPTG (Ptac) or 0.1% rhamnose (T7), respectively. Afterwards, the incubation temperature was reduced to 20°C for protein expression. Cells were harvested and proteins were extracted via chemical cell destruction. A standard SDS-PAGE was used to separate the proteins. Afterwards, the proteins in the desired size ranges were extracted and digested by trypsin. Finally, MALDI-TOF-MS was applied to identify the peptide fragments.
Isobutanol production and quantification
E. coli with BBa_K1465306 and BBa_K1465307 was grown in LB media with 30 µg/mL chloramphenicol for the production of isobutanol. Samples for both constructs were taken at different time points during the cultivation. To identify the optimal cultivation temperature two separate cultivations were performed at 37°C and 30°C, respectively. GC-MS was applied for the isobutanol quantification in the supernatant of the cultures. Finally, the isobutanol production was calculated based on a calibration curve.
The loss of isobutanol due to evaporation was studied by comparing flasks that were opened with different frequencies. All flasks started with the same initial amount of isobutanol and were incubated at 37°C over 20 h. One group was opened 10 times during incubation while the other flasks were only opened once at the end. Afterwards, isobutanol was quantified via GS-MS as described before.
Antibiotic-free selection system
Our antibiotic-free selection plasmid (BBa_K1465401) encoding the alanine racemase (Alr) was transformed in different concentrations into E. coli DH5α Δalr ΔdadX. After regeneration the cells were streaked out either onto LB plates for the antibiotic-free selection via complementation of the D-alanine auxotrophy or onto LB plates containing 30 µg/mL chloramphenicol and 3 mM D-alanine as a control, respectively. Successful transformation events could be observed by the red color of colonies caused by the reporter RFP, while false-positive colonies remained white.
Results and Discussion
Electron transfer
We engineered an E. coli strain to accept electrons stimulating its metabolism by focusing on an indirect electron transfer via a mediator. This electron transfer system consists of multiple steps. First, the reduced mediator has to cross the outer membrane of the E. coli cell wall. After crossing the periplasmatic space, the mediator adsorbs at the inner membrane of the E. coli cell wall and functions as an electron donor for the membrane associated fumarate reductase. In this step succinate is produced in the cytoplasm by the reduction of fumarate into succinate. Under oxygen limitation the C4 carboxylate transporter DcuB is responsible for the fumarate import and succinate export. To avoid the succinate export in our project, this antiporter was deleted. The generation of succinate creates a loop into the citric acid cycle, because succinate is reoxidized again by the succinate dehydrogenase. The succinate dehydrogenase catalyzes the transfer of electrons to FAD+ generating FADH2, which enters the electron transport chain. The electron transport facilitates proton translocation over the inner bacterial membrane. The proton motoric force is used by ATP synthase. Generated ATP and reductive power in the bacterial cell lead to an increasing metabolic activity.
In order to allow the mediator to cross the outer membrane, the outer membrane porine encoding gene oprF (BBa_K1172507) from Pseudomonas fluorescens was expressed in E. coli. Therefore, oprF was integrated in the bacterial chromosome by replacing dcuB, which encodes the C4 carboxylate transporter. The successful knockout of dcuB and simultaneous insertion of oprF was shown with PCR analysis and DNA sequencing. The functionality of the outer membrane porin OprF in E. coli KRX ΔdcuB::oprF was investigated via NPN-Uptake-Assay [29]. 1‑N‑Phenylnaphthylamine (NPN) changes fluorescence activity between aqueous and hydrophobic milieus. There is only minor fluorescence in aqueous solution, but the transport in the hydrophobic periplasmatic space causes an increased fluorescence. So NPN fluorescence is a good indicator for membrane permeability [30]. An increased amount of the membrane porine OprF leads to an increasing membrane permeability. The results are shown in Fig. 1. Compared to E. coli KRX wild type an increased fluorescence emission can be observed for oprF expressing strains and an advanced membrane permeability can be demonstrated for these strains. The successful deletion of dcuB was also shown with phenotypic investigation via Biolog analysis and anaerobic cultivation in M9 minimal media with fumarate supplemented. The results are shown in Fig. 1. The results of the anaerobic cultivation of E. coli KRX ΔdcuB::oprF showed no excretion of succinate in contrast to the E. coli KRX wild type. This is caused by the deletion of the antiporter gene dcuB, which is responsible for fumarate uptake and succinate export under anaerobic or oxygen-limiting conditions.
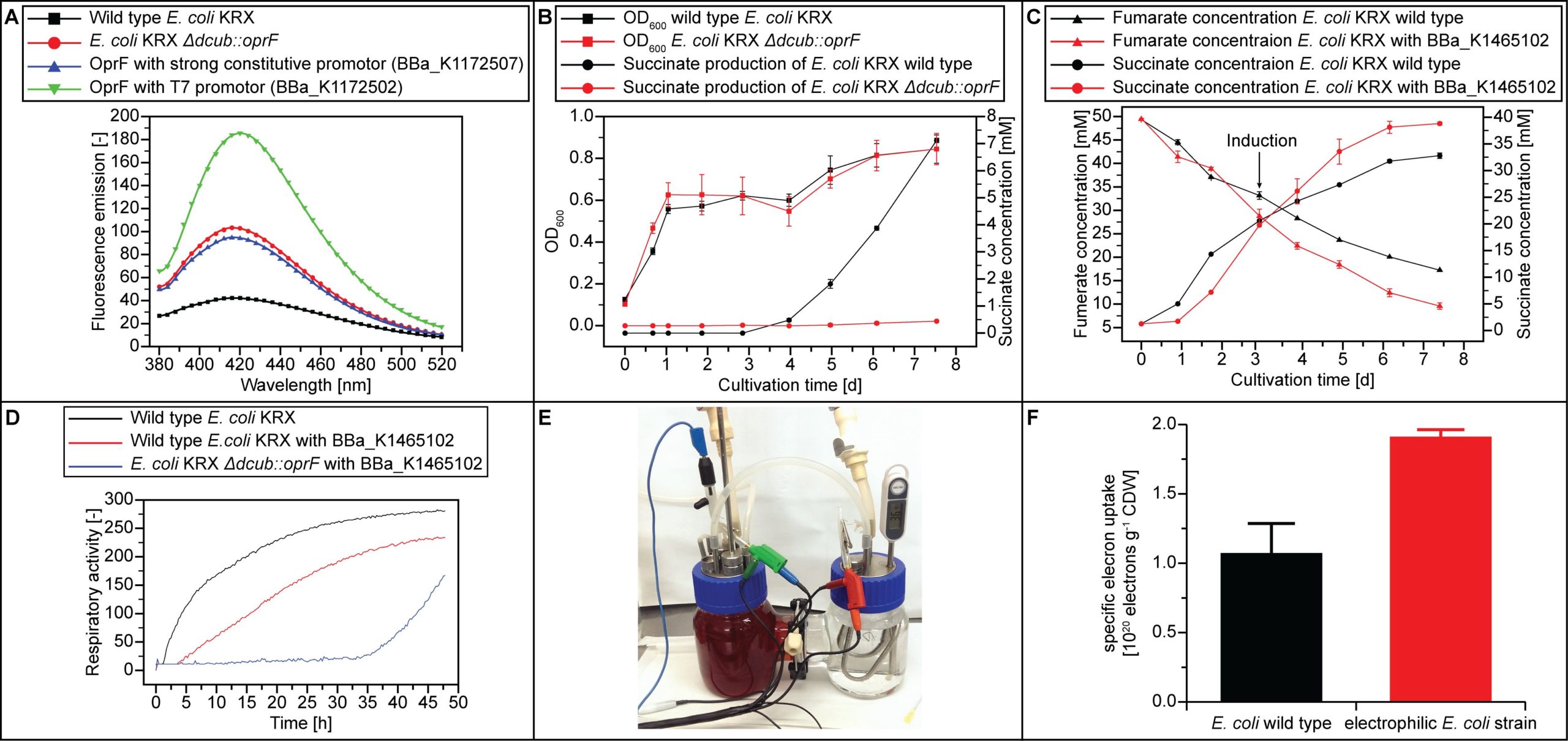
The next step of the electron uptake system is the membrane associated fumarate reductase that converts fumarate into succinate by transferring electrons from the mediator in the periplasmatic space on fumarate. The fumarate reductase gene frd was overexpressed under the control of the T7 promoter. The expression was shown via SDS-PAGE and MALDI-TOF-MS analysis. Functionality was tested by anaerobic cultivation in M9 minimal medium with fumarate supplemented (Fig. 1). Growth and succinate production was compared to E. coli KRX wild type. In the case of fumarate in the medium fumarate respiration takes place under anaerobic conditions [31]. About 72 hours after induction of the cells, the impact of frd expression becomes clear. The succinate production increased and reached a higher final concentration compared to the wild type with similar growth. Furthermore, an increased fumarate consumption could be observed in the frd overexpressing strain after induction. This shows functionality of the fumarate reductase in E. coli KRX. These findings were confirmed with a Biolog experiment.
Finally, the fumarate reductase frd was expressed in E. coli KRX ΔdcuB::oprF to increase the electron uptake. The electro biochemical behavior of E. coli KRX with deleted C4 carboxylate antiporter DcuB, expressed porin OprF and overexpressed fumarate reductase frd was shown in an H-cell reactor (Fig. 1). In this experiment an increase electron uptake of 72% compared to E. coli KRX wild type has been observed.
Calvin cycle in E. coli
As previously mentioned, E. coli lacks the three enzymes SBPase, PrkA and RuBisCO to enable a functional Calvin cycle. To close the first gap in the cycle, the SBPase (GlpX) from Bacillus methanolicus [27] was chosen as it is one of the best characterized enzymes for this function. Therefore, the protein coding sequence of glpX was cloned into pSB1C3 (BBa_K1465228). SBPase activity was validated in an enzyme assay (Fig. 2). Hence, a comparison between different temperatures (37°C and 50°C) in an in vitro assay was performed to investigate the different enzymatic activities to show the feasibility for E. coli (Fig. 2). SBPase activity was observed at 37°C but the activity was higher at 50°C. The observed difference in the SBPase activity between 37°C and 50°C is probably due to its origin from the thermophilic B. methanolicus [32].
To add the second enzyme function, the prkA coding sequence obtained from Synechococcus elongatus was synthesized to remove restriction sites incompatible with the iGEM standard and to optimize the codon usage for E. coli (BBa_K1465201). Expression of prkA in E. coli via BBa_K1465212 leads to an accumulation of Ru-BP that cannot be further metabolized and is therefore cell toxic [12,33]. A protein extract of this expression strain loaded onto a SDS-PAGE revealed a band between 35 kD and 40 kD, representing the PrkA that has a molecular size of approximately 38 kD. MALDI-TOF-MS analysis of the extracted and digested PrkA identified three peptides. However, it was not possible to show the enzyme functionality in an in vitro assay. A possible explanation is the light dependent activation of the PrkA in plants. The light triggers the photo system I in photosynthetic active organisms. These triggers reduce ferredoxine. In the following reaction thioredoxin is reduced while ferredoxin is oxidized again. The reduced thioredoxin is able to break disulfides. The PrkA activation is triggered by thioredoxin that might not be sufficiently present in E. coli. It would be possible to activate the PrkA by adding DTT to the enzyme assay [34].
Finally, the coding sequence of the actual carbon dioxide fixing enzyme, the RuBisCO, was cloned either under the control of a Ptac (BBa_K1465213) or a T7 promoter. The presence of both subunits were confirmed using SDS-PAGE and MALDI-TOF-MS. RuBisCO activity was validated via an in vitro assay measuring the consumption of Ru-BP and the production of PGA. These two metabolites are the substrate and product of the RuBisCO, respectively. The substances are clearly separable via HPLC with a retention time of 14.4 min for Ru-BP and 12.6 min for 3-PGA, which was verified by using comparable standards (Fig 3). Cell extracts of KRX wild type and KRX carrying the construct BBa_K1465202 were compared over time (Fig. 3). No change of the PGA peak was observed in the wild type cell extract after Ru-BP was added. However, the cell extract of our engineered strain showed RuBisCO activity by the consumption of Ru-BP and the production of PGA. It appears that the reaction is not completed after 15 min, because there is still Ru-BP detectable. This may be due to the low reaction rate and high KM value of the RuBisCO [35]. However, a number of cellular reactions yields PGA. This explains the small amount of PGA in both cell extracts. Nevertheless, conversion of Ru-BP to PGA is a specific reaction catalyzed by RuBisCO. This is confirmed by the lack of PGA accumulation in the wild type cell extract containing Ru-BP but no RuBisCO. Finally, it can be concluded that the RuBisCO is functionally expressed and active, showing the highly specific conversion of Ru-BP to PGA over time. The comprehensive characterization of the RuBisCO could be further improved by utilizing 14C marked substances [36–38].

Protein-based microcompartment functions as carboxysome
As a bacterial microcompartment, which could be used as a local enrichment atmosphere of CO2 we further investigated the carboxysome from H. neapolitanus [39]. We provided different subunits of the carboxysome (BBa_K1465202 – BBa_K1465223) and further investigated the codon optimized synthetic carboxysome (BBa_K1465223). It encodes the shell proteins CsoS1ABC and CsoS4AB as well as the shell associated protein CsoS2. A bottom up construction approach enabled the identification of the essential parts. We used a translational fusion of one of the sequences encoding the shell proteins CsoS1A and GFP (BBa_K1465222) as an indicator of correct protein folding. A concentrated subcellular localization of the GFP fluorescence shows the positions of carboxysomes within the cells (Fig. 4). This reporter function of GFP was identified and used for this purpose before [40,41]. Additionally, we analyzed the functions of different shell proteins with incomplete functional characterization [42]. The assembly of the carboxysome was achieved without the expression of csoS1D, another coding sequence that is located on the plasmid pHnCBS1D. The resulting CsoS1D is probably responsible for the pore size in the carboxysome shell [12,43]. Nevertheless, it seems that it has no essential function in protein folding.
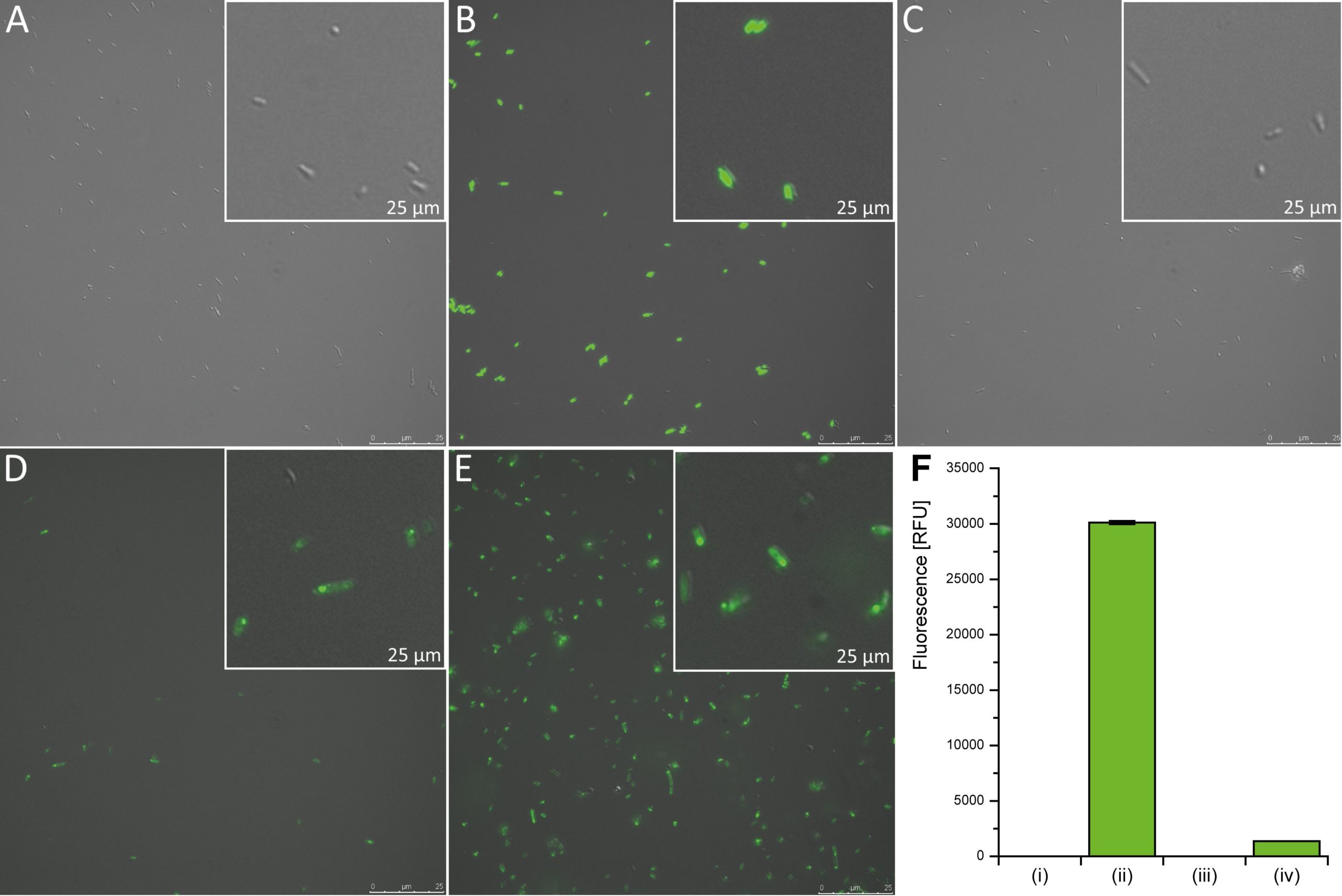
Fluorescence of GFP fusion proteins was quantified by photometric measurements (Fig. 4). The signal intensity of the E. coli wild type (negative control) is compared to cells carrying different constructs. E. coli cells constitutively expressing gfp under the Ptet promoter served as a positive control and lead to the highest fluorescence signal. The expression of csoS1A::gfp together with the shell protein encoding genes csoS1BC and csoS4AB (BBa_K1465222) did not yield any fluorescence. However, the combined expression of csoS2 and the shell protein encoding genes csoS1A::gfp-csoS1BC and csoS4AB (BBa_K1465223) results in detectable green fluorescence. From these results we deduce that the correct folding of the shell proteins and the assembly of the whole carboxysome is dependent on the presence of the shell associated protein CsoS2.
High resolution microscopy revealed the subcellular localization of carboxysomes within the cells (Fig. 4). Most of the cells seem to have only one carboxysome, but there are also some cells that carry two. Additionally, we could detect some cells forming a filament located at the membrane of the cell (Fig. 4). These filaments might be formed because of a high gene expression of the plasmid [12]. The T7 promoter could explain this observation due to the high transcription rate of the T7 polymerase. Estimation of the carboxysome size resulted in about 0.3 µm for the entire protein structure. This value is almost double the size compared to previous works that reported a size of approximately 0.136 µm (+/- 0.036) [12]. Our BioBricks might be useful to future iGEM projects focusing on other oxygen sensitive reactions like nitrogen fixation.
Isobutanol production pathway
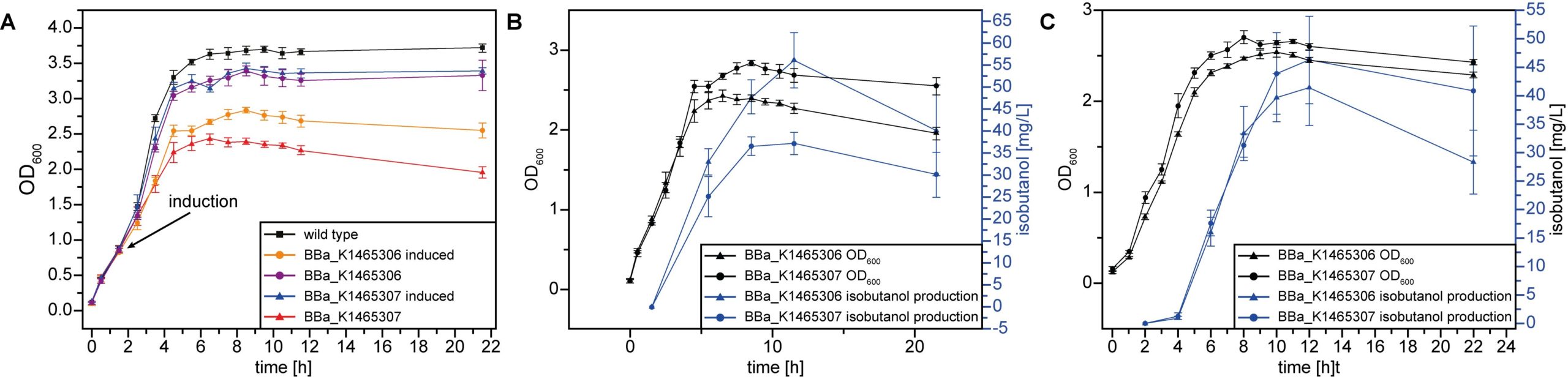
Five enzymatically catalyzed steps are required to produce isobutanol from pyruvate in E. coli [16]. We used existing BioBricks containing the coding sequences for the α‑acetolactate synthase (AlsS) of Bacillus subtilis, the ketol-acid reductoisomerase (IlvC) and Dihydroxyacid dehydratase (IlvD) of E. coli and the α-ketoisovalerate decarboxylase (KivD) of Lactococcus lactis to construct the plasmid BBa_K1465302. The last step of the isobutanol production pathway is catalyzed by the endogenous alcohol dehydrogenase of E. coli (AdhE). Additionally, we tested the alcohol dehydrogenase (adhA) of L. lactis by adding it to BBa_K1465302 to further enhance the isobutanol production (BBa_K1465303). This alcohol dehydrogenase was identified as an alternative enzyme for the last step in the 2-keto-acid pathway that is responsible for the isobutanol production [44]. For cultivation experiments the combined coding sequences were placed under control of the Ptac promoter (BBa_K731500) resulting in the plasmids BBa_K1465306 and BBa_K1465307, respectively.
Cultivation experiments were performed to quantify the isobutanol production of BBa_K1465306 and BBa_K1465307 carrying strains. A reduced growth for the bacteria expressing the genes of the plasmids compared to the wild type or the uninduced control was observed (Fig. 5). This was probably due to the high burden of additional protein biosynthesis. Moreover, both introduced pathway versions (BBa_K1465306 and BBa_K1465307) led to a production of 13 to 40 mg/L isobutanol within 20 hours depending on the specific cultivation conditions (Fig. 5).
To quantify the impact of isobutanol evaporation during the sampling process, the amount of isobutanol in flasks opened ten times and flasks opened only at the end of the experiment was compared. However, no significant difference was found between the differentially treated flasks as about 50% isobutanol was lost in both cases.
Antibiotic-free selection
One important part of iGEM is human practices. Participants consider different aspects of their project and possible impacts on humanity and the environment. We identified the frequent use of antibiotics as a major issue and started the construction of an antibiotic-free selection system for the iGEM community. Moreover, when thinking of an application of the project outside the laboratory, it is crucial to keep antibiotic resistance from spreading out.
The double deletion of the constitutive alanine racemase (alr) and the catabolic alanine racemase (dadX) in both, the E. coli DH5α and KRX strains, resulted in the genotypes KRX Δalr ΔdadX and DH5α Δalr ΔdadX, respectively. Both strains showed a strict D-alanine auxotrophy that could be complemented by our plasmid (BBa_K1465401). This plasmid carries the constitutively expressed alanine racemase. Additionally, this plasmid contains the coding sequence of a chloramphenicol acetyltransferase for chloramphenicol-resistance and RFP (BBa_J04450) as a reporter to distinguish between successful transformated cells and false positives.
The comparison between the chloramphenicol selection and the antibiotic-free selection revealed a threefold difference in transformation efficiency favoring the antibiotic-free selection system. Comparable results were obtained for different plasmid concentrations leading to the assumption that chloramphenicol inhibits the early cell formation, while a lack of D-alanine within the antibiotic-free selection can be compensated by intracellular storage of D-alanine. This is supported by the observation that even an antibiotic-free selection without any D-alanine complementation results in some countable colonies. However, it should also be mentioned that the rate of reversion is about 3.27*10-7 ± 2.27*10-7, which might be due to a point mutation of metJ [45]. Although the experiments were performed with the relatively small plasmid BBa_K1465401 (4.2 kb) and might be even more challenging for more complicated cloning approaches, it did not escape our notice that this antibiotic-free selection system is feasible for other bacteria where the peptidoglycan is also cross-linked by D-alanine, such as Listeria monocytes [46], Corynebacterium glutamicum [47], or B. subtilis [48].
Conclusion
We designed and tested different setups in order to cultivate and analyze the constructed electrophilic E. coli strain. Further experiments will determine whether the electron uptake of the electrophilic strain can power the reconstructed Calvin cycle in E. coli. Although we were able to verify the function of all missing parts of the whole Calvin cycle separately, it remains challenging to verify their combined function as well as inside the electrophilic strain. Additionally, further characterization of the RuBisCO integrated in the carboxysome will reveal if the anaerobic conditions will increase the efficiency of the carbon dioxide fixation. Moreover, we constructed and validated a synthetic pathway for the production of isobutanol based on BioBricks. Unfortunately, the final combination of all subparts of the project could not be shown during the competition. Nevertheless, confirmed BioBricks for biofuel production and microcompartments with anaerobic conditions will be very useful to future iGEM teams.
Finally, we demonstrated an initial step for a real world application of the system by establishing an antibiotic-free selection plasmid. This selection system is now available to the whole iGEM community and could contribute to a significant reduction of antibiotic use in future synthetic biology projects.
Acknowledgements
We thank all members of the Center for Biotechnology (CeBiTec) and Bielefeld University who supported us and our project. In particular, Prof. Dr. Jörn Kalinowski and Dr. Christian Rückert (Research Group Microbial Genomics and Biotechnology at CeBiTec, Bielefeld University) and Prof. Dr. Kristian Müller (Research Group Cellular and Molecular Biotechnology at the Technical Faculty, Bielefeld University) for their extensive support during our participation in iGEM. Moreover, we would like to thank all external supporters of our project.
Response to Reviewers
A transcript of the reviewer comments and author responses from the Live Peer Review Jamboree can be found here: Bielefeld 2014 Response to Reviewers
References
- Lin L, Cunshan Z, Vittayapadung S, Xiangqian S, Mingdong D. Opportunities and challenges for biodiesel fuel. Appl Energy. 2011;88: 1020–1031. doi:10.1016/j.apenergy.2010.09.029
- Rahman F, Rehman S, Abdul-Majeed MA. Overview of energy storage systems for storing electricity from renewable energy sources in Saudi Arabia. Renew Sustain Energy Rev. 2012;16: 274–283. doi:10.1016/j.rser.2011.07.153
- Lovley DR. Powering microbes with electricity: direct electron transfer from electrodes to microbes. Environ Microbiol Rep. 2011;3: 27–35. doi:10.1111/j.1758-2229.2010.00211.x
- Krieg T, Sydow A, Schröder U, Schrader J, Holtmann D. Reactor concepts for bioelectrochemical syntheses and energy conversion. Trends Biotechnol. 2014;32: 645–655. doi:10.1016/j.tibtech.2014.10.004
- Park DH, Laivenieks M, Guettler MV, Jain MK, Zeikus JG. Microbial Utilization of Electrically Reduced Neutral Red as the Sole Electron Donor for Growth and Metabolite Production. Appl Environ Microbiol. 1999;65: 2912–2917.
- D’Alessandro DM, Smit B, Long JR. Carbon Dioxide Capture: Prospects for New Materials. Angew Chem Int Ed. 2010;49: 6058–6082. doi:10.1002/anie.201000431
- Fongwa E, Onyango V, Gnauck A. Review of Future Energy Supply and Targets for Climate Change: The Idea of Ecosystem Services. In: Schmidt M, Onyango V, Palekhov D, editors. Implementing Environmental and Resource Management. Springer Berlin Heidelberg; 2011. pp. 119–132. doi:10.1007/978-3-540-77568-3_12
- Sumida K, Rogow DL, Mason JA, McDonald TM, Bloch ED, Herm ZR, et al. Carbon Dioxide Capture in Metal–Organic Frameworks. Chem Rev. 2012;112: 724–781. doi:10.1021/cr2003272
- Leytem AB, Dungan RS, Bjorneberg DL, Koehn AC. Emissions of Ammonia, Methane, Carbon Dioxide, and Nitrous Oxide from Dairy Cattle Housing and Manure Management Systems. J Environ Qual. 2011;40: 1383. doi:10.2134/jeq2009.0515
- Baccini A, Goetz SJ, Walker WS, Laporte NT, Sun M, Sulla-Menashe D, et al. Estimated carbon dioxide emissions from tropical deforestation improved by carbon-density maps. Nat Clim Change. 2012;2: 182–185. doi:10.1038/nclimate1354
- Mattozzi M d, Ziesack M, Voges MJ, Silver PA, Way JC. Expression of the sub-pathways of the Chloroflexus aurantiacus 3-hydroxypropionate carbon fixation bicycle in E. coli: Toward horizontal transfer of autotrophic growth. Metab Eng. 2013;16: 130–139. doi:10.1016/j.ymben.2013.01.005
- Bonacci W, Teng PK, Afonso B, Niederholtmeyer H, Grob P, Silver PA, et al. Modularity of a carbon-fixing protein organelle. Proc Natl Acad Sci. 2012;109: 478–483. doi:10.1073/pnas.1108557109
- Zhuang Z-Y, Li S-Y. Rubisco-based engineered Escherichia coli for in situ carbon dioxide recycling. Bioresour Technol. 2013;150: 79–88. doi:10.1016/j.biortech.2013.09.116
- Osmond CB. Photorespiration and photoinhibition. Biochim Biophys Acta BBA – Rev Bioenerg. 1981;639: 77–98. doi:10.1016/0304-4173(81)90006-9
- Bassham JA, Benson AA, Kay LD, Harris AZ, Wilson AT, Calvin M. The Path of Carbon in Photosynthesis. XXI. The Cyclic Regeneration of Carbon Dioxide Acceptor1. J Am Chem Soc. 1954;76: 1760–1770. doi:10.1021/ja01636a012
- Atsumi S, Hanai T, Liao JC. Non-fermentative pathways for synthesis of branched-chain higher alcohols as biofuels. Nature. 2008;451: 86–89. doi:10.1038/nature06450
- Karabektas M, Hosoz M. Performance and emission characteristics of a diesel engine using isobutanol–diesel fuel blends. Renew Energy. 2009;34: 1554–1559. doi:10.1016/j.renene.2008.11.003
- Peralta-Yahya PP, Zhang F, del Cardayre SB, Keasling JD. Microbial engineering for the production of advanced biofuels. Nature. 2012;488: 320–328. doi:10.1038/nature11478
- Bala JD, Lalung J, Al-Gheethi AAS, Norli I. A Review on Biofuel and Bioresources for Environmental Applications. In: Ahmad MI, Ismail M, Riffat S, editors. Renewable Energy and Sustainable Technologies for Building and Environmental Applications. Springer International Publishing; 2016. pp. 205–225. doi:10.1007/978-3-319-31840-0_13
- Wright O, Stan G-B, Ellis T. Building-in biosafety for synthetic biology. Microbiol Read Engl. 2013;159: 1221–1235. doi:10.1099/mic.0.066308-0
- Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mpl8 and pUC19 vectors. Gene. 1985;33: 103–119. doi:10.1016/0378-1119(85)90120-9
- de Lorenzo V, Herrero M, Jakubzik U, Timmis KN. Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J Bacteriol. 1990;172: 6568–6572.
- Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, et al. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene. 1995;166: 175–176.
- Chopra I, Roberts M. Tetracycline Antibiotics: Mode of Action, Applications, Molecular Biology, and Epidemiology of Bacterial Resistance. Microbiol Mol Biol Rev. 2001;65: 232–260. doi:10.1128/MMBR.65.2.232-260.2001
- Walsh CT. Enzymes in the D-alanine branch of bacterial cell wall peptidoglycan assembly. J Biol Chem. 1989;264: 2393–2396.
- iGEM Bielefeld-CeBiTec 2014. Bielefeld-CeBiTec 2014 Protocols. In: Bielefeld-CeBiTec 2014 [Internet]. 2014 [cited 10 Oct 2016]. Available: http://2014.igem.org/Team:Bielefeld-CeBiTec/Notebook/Protocols
- Stolzenberger J, Lindner SN, Persicke M, Brautaset T, Wendisch VF. Characterization of fructose 1,6-bisphosphatase and sedoheptulose 1,7-bisphosphatase from the facultative ribulose monophosphate cycle methylotroph Bacillus methanolicus. J Bacteriol. 2013;195: 5112–5122. doi:10.1128/JB.00672-13
- Chakrabarti S, Bhattacharya S, Bhattacharya SK. A nonradioactive assay method for determination of enzymatic activity of d-ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco). J Biochem Biophys Methods. 2002;52: 179–187. doi:10.1016/S0165-022X(02)00072-6
- Tzeng SR, Cheng J wai. New antibiotic peptides, useful in treating or preventing a microbial or viral infections or in inactivating Gram-positive and -negative bacteria, protozoa, fungi and virus [Internet]. Available: http://www.google.com/patents/DE10360435A1
- Loh B, Grant C, Hancock RE. Use of the fluorescent probe 1-N-phenylnaphthylamine to study the interactions of aminoglycoside antibiotics with the outer membrane of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1984;26: 546–551.
- Iverson TM, Luna-Chavez C, Cecchini G, Rees DC. Structure of the Escherichia coli Fumarate Reductase Respiratory Complex. Science. 1999;284: 1961–1966. doi:10.1126/science.284.5422.1961
- Brautaset T, Jakobsen ØM, Josefsen KD, Flickinger MC, Ellingsen TE. Bacillus methanolicus: a candidate for industrial production of amino acids from methanol at 50°C. Appl Microbiol Biotechnol. 2007;74: 22–34. doi:10.1007/s00253-006-0757-z
- Parikh MR, Greene DN, Woods KK, Matsumura I. Directed evolution of RuBisCO hypermorphs through genetic selection in engineered E.coli. Protein Eng Des Sel PEDS. 2006;19: 113–119. doi:10.1093/protein/gzj010
- Hariharan T, Johnson PJ, Ann Cattolico R. Purification and Characterization of Phosphoribulokinase from the Marine Chromophytic Alga Heterosigma carterae. Plant Physiol. 1998;117: 321–329.
- Sage RF. Variation in the k(cat) of Rubisco in C(3) and C(4) plants and some implications for photosynthetic performance at high and low temperature. J Exp Bot. 2002;53: 609–620.
- Butz ND, Sharkey TD. Activity Ratios of Ribulose-1,5-Bisphosphate Carboxylase Accurately Reflect Carbamylation Ratios 1. Plant Physiol. 1989;89: 735–739.
- Sage RF, Reid CD, Moore B d, Seemann JR. Long-term kinetics of the light-dependent regulation of ribulose-1,5-bisphosphate carboxylase/oxygenase activity in plants with and without 2-carboxyarabinitol 1-phosphate. Planta. 191: 222–230. doi:10.1007/BF00199753
- Sage RF, Seemann JR. Regulation of Ribulose-1,5-Bisphosphate Carboxylase/Oxygenase Activity in Response to Reduced Light Intensity in C4 Plants. Plant Physiol. 1993;102: 21–28.
- Cannon GC, Shively JM. Characterization of a homogenous preparation of carboxysomes from Thiobacillus neapolitanus. Arch Microbiol. 1983;134: 52–59. doi:10.1007/BF00429407
- Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol. 1999;17: 691–695. doi:10.1038/10904
- Hsu S-TD, Blaser G, Jackson SE. The folding, stability and conformational dynamics of beta-barrel fluorescent proteins. Chem Soc Rev. 2009;38: 2951–2965. doi:10.1039/b908170b
- Yeates TO, Kerfeld CA, Heinhorst S, Cannon GC, Shively JM. Protein-based organelles in bacteria: carboxysomes and related microcompartments. Nat Rev Microbiol. 2008;6: 681–691. doi:10.1038/nrmicro1913
- Kinney JN, Axen SD, Kerfeld CA. Comparative analysis of carboxysome shell proteins. Photosynth Res. 2011;109: 21–32. doi:10.1007/s11120-011-9624-6
- Atsumi S, Wu T-Y, Eckl E-M, Hawkins SD, Buelter T, Liao JC. Engineering the isobutanol biosynthetic pathway in Escherichia coli by comparison of three aldehyde reductase/alcohol dehydrogenase genes. Appl Microbiol Biotechnol. 2010;85: 651–657. doi:10.1007/s00253-009-2085-6
- Kang L, Shaw AC, Xu D, Xia W, Zhang J, Deng J, et al. Upregulation of MetC Is Essential for d-Alanine-Independent Growth of an alr/dadX-Deficient Escherichia coli Strain. J Bacteriol. 2011;193: 1098–1106. doi:10.1128/JB.01027-10
- Thompson RJ, Bouwer HGA, Portnoy DA, Frankel FR. Pathogenicity and Immunogenicity of a Listeria monocytogenes Strain That Requires d-Alanine for Growth. Infect Immun. 1998;66: 3552–3561.
- Tauch A, Götker S, Pühler A, Kalinowski J, Thierbach G. The alanine racemase gene alr is an alternative to antibiotic resistance genes in cloning systems for industrial Corynebacterium glutamicum strains. J Biotechnol. 2002;99: 79–91.
- Ferrari E, Henner DJ, Yang MY. Isolation of an Alanine Racemase Gene from Bacillus subtilis and its Use for Plasmid Maintenance in B. subtilis. Nat Biotechnol. 1985;3: 1003–1007. doi:10.1038/nbt1185-1003